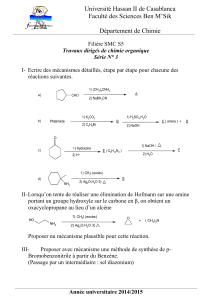
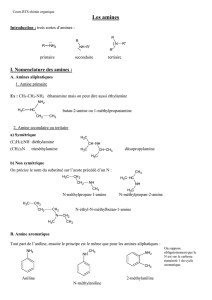
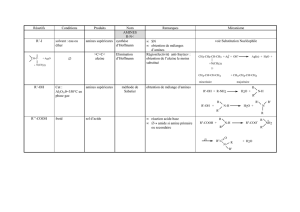
2 Les amines

30
2 Les amines
Les amines sont des dérivés de l’ammoniac (NH3) et elles sont très abondantes
dans la nature (protéines, peptides, acides aminés, alcaloïdes, etc.). Les amines sont dites
primaires, secondaires ou tertiaires si l’atome d’azote est substitué par un (primaire),
deux (secondaire) ou trois (tertiaire) substituants carbonés comme il est indiqué à la suite
(figure 2-1).
R-NH2 R
2NH R3N R4N+X-
amine primaire amine secondaire amine tertiaire sel d’ammonium
quaternaire
Figure 2-1 : Classes de fonctions amines.
Certains dérivés des amines manifestent des activités physiologiques importantes
comme le témoignent les molécules représentées à la suite (figure 2-2). Un bon nombre
de ces produits semblent agir sur des récepteurs du cerveau (amphétamine, mescaline,
ecstasy, morphine, codéine, héroïne, adrénaline). Certaines similitudes structurales entre
ces produits permettent de croire que la présence d’une fonction amine séparée du noyau
aromatique par deux carbones soit un élément de design important pour l’interaction avec
les récepteurs du cerveau.
Il est intéressant de rappeler que les amines ont un arrangement moléculaire
tétraédrique similaire à un carbone sp3. Trois des sommets du tétraèdre sont occupés par
les substituants tandis que le quatrième est occupé par une paire d’électrons libres. Cet
arrangement tétraédrique autour de l’azote permet de penser que ces molécules sont
chirales si les trois substituants sont différents. Un tel composé devrait exister sous la
forme de deux énantiomères possédant un pouvoir rotatoire propre (après séparation des
deux énantiomères). En fait, les amines simples constituées de trois substituants différents
n’ont jamais été isolé pour la simple raison que ces composés s’isomérisent très
rapidement et facilement par un phénomène d’inversion (inversion de l’azote). Cette
inversion ne nécessite pas l’intervention d’un réactif extérieur. On peut concevoir que la
molécule passe par un état de transition où l’azote est hybridé en sp2 comme il est montré
à la suite (figure 2-3).

31
OH
OH
HO
NHCH
3
H3CO
OCH3
OCH3
NH2
O
OHN
Adrénaline Mescaline Ecstasy
O
R
1
O
R
2
O
NCH
3
H
Cl
N
CH
3
CH
3
CH
3
CH
3
NH
2
CH
3
Morphine (R1 = R2 = H)
Héroïne (R1 = R2 = COCH3)
Codéine (R1 = CH3 et R2 =
H)
Méridia
(sibutramine)
coupe-faim
Amphétamine
Figure 2-2 : Exemple de composés aminés bioactifs.

32
N
C
2
H
5
CH
3
H
NC
2
H
5
H
3
CH
N
C
2
H
5
CH
3
H
N
C
2
H
5
CH
3
H
C
2
H
5
NCH
3
H
Figure 2-3 : Inversion des amines.
Il est intéressant de préciser que les sels d’ammonium constituées de quatre
substituants différents sont chiraux et relativement stables (figure 2-4). La barrière
d’énergie d’inversion du stéréocentre séparant les deux énantiomères est plus grande chez
les sels d’ammonium que pour les amines ordinaires.
CH3
NC6H5
CH2
C2H5
CHCH2
CH3
N
C6H5CH2
C2H5
CH CH2
Figure 2-4 : Amines quaternaires optiquement actives.
Les amines se comportent comme des bases (la paire d’électrons libres est utilisée
pour effectuer une protonation). Le caractère basique est relativement marqué (pKb de 4
pour les amines primaires aliphatiques). Les amines peuvent également jouer le rôle de
nucléophiles (plus fort qu’un alcool).
2.1 Basicité des amines aromatiques
La basicité des amines aromatiques est influencée par les substituants présents sur
le noyau aromatique (tableau 2-1). D’une façon générale, les groupements activants
(comme -OCH3, -NH2, -CH3, etc.) augmentent le caractère basique d’une amine
aromatique tandis que les groupements désactivants (groupements carbonyles, nitro, etc.)
diminuent le caractère basique d’une amine aromatique (figure 2-5). Les groupements
activants (électrodonneur par effet inductif ou par résonance) augmentent la densité
électronique sur la fonction amine. D’un autre point de vue, les groupements activants
exercent une influence stabilisante sur le produit de protonation comme le montre
l’exemple à la suite. Dans le cas des groupements désactivants, ils diminuent la densité
électronique sur l’amine et ainsi son caractère basique.

33
Tableau 2-1 : Quelque pKb des dérivés de l’aniline
Dérivés pKb
Aniline 9.4
p-bromoaniline 10.1
m-cyanoaniline 11.3
p-nitroaniline 13
p-méthoxyaniline 8.7
NH
3
Z
NH
3
R
Effet déstabilisant
Bases moins fortes
Effet stabilisant
Bases plus fortes
Figure 2-5 : Effet des substituants sur la basicité de l’aniline.
2.2 Préparation des amines
Les amines peuvent être préparées en utilisant plusieurs approches synthétiques.
Cette section met brièvement en évidence quelques méthodes bien connues pour la
préparation des amines.
2.2.1 Alkylation des amines
Les amines sont des nucléophiles et elles réagissent avec les halogénoalcanes pour
donner des sels d’ammonium. Les rendements de ces réactions sont faibles car le produit
de la première alkylation est souvent impliqué dans des réactions d’alkylation

34
subséquentes. L’alkylation des amines par cette approche entraîne la formation d’un
mélange complexe de sels d’ammonium et d’alkylamines comme le montre la série de
réactions suivantes (figure 2-6).
NH
3
CH
3
Br+CH
3
NH
3
Br+
CH
3
NH
3
NH
3
+CH
3
NH
2
NH
4
+
CH
3
NH
2
CH
3
Br+(CH
3
)
2
NH
2
Br+
(CH
3
)
2
NH
2
NH
3
+(CH
3
)
2
NH NH
4
+
(CH
3
)
2
NH CH
3
Br+(CH
3
)
3
NH Br+
(CH
3
)
3
NH NH
3
+(CH
3
)
3
NNH
4
+
Figure 2-6 : Alkylation des amines par substitution nucléophile sur les halogénoalcanes.
Exercice 21-6 (p. 950)
2.2.2 Préparation des amines primaires via l’ion cyanure et la synthèse de Gabriel
L’alkylation contrôlée (pour éviter la polyalkylation) des amines, implique
l’emploi d’un nucléophile contenant de l’azote et qui peut réagir qu’une seule fois. L’ion
cyanure (-CN) transforme les halogénoalcanes en nitriles (via une substitution
nucléophile), qui peuvent par la suite être réduits en amines (figure 2-7). De cette façon,
il est possible de préparer une amine primaire contenant un atome de carbone
supplémentaire par rapport au dérivé halogéné (R-X vers R-CH2-NH2).
RX CN RCN
+X
RCN H
2
catalyseur RCH
2
NH
2
Figure 2-7 : Préparation d’amine primaire via une réaction de cyanation.
La synthèse de Gabriel peut également être utilisée pour préparer des amines
primaires et ce, sans être contraint de faire usage d’une réaction de réduction. Pour ce
faire, il est nécessaire d’utiliser un imide cyclique comme le phthalimide. Ce dernier se
substitut au chlore sur le groupement d’intérêt pour être ensuite hydrolysé en condition
basique puis, acidifié à la fin (figure 2-8).
 6
7
8
9
10
11
12
13
14
6
7
8
9
10
11
12
13
14
1
/
14
100%