Le cycle cellulaire : données biologiques et thérapies ciblant les

Correspondances en Onco-Théranostic - Vol. VI - n° 1 - janvier-février-mars 2017
30
dossier thématique
Prolifération
et cycle cellulaire
Le cycle cellulaire : données biologiques
et thérapies ciblant les cyclines/CDK
Cell cycle: biological data and treatment targeting cyclins/CDKs
O. Trédan1, 2, P. Barthelemy3, C. Villanueva4, L. Teixeira5
1 Département de
cancérologie médicale,
centre Léon-Bérard, Lyon.
2 CNRS UMR5286,
Centre de recherche en
cancérologie de Lyon
(CRCL).
3 Unité d’oncologie
médicale, hôpitaux
universitaires de
Strasbourg, Institut
régional du cancer
d’Alsace, Strasbourg.
4 Service d’oncologie
médicale, centre
hospitalier universitaire
deBesançon.
5 Centre des maladies du
sein, service d’oncologie
médicale, hôpital Saint-
Louis (AP-HP), Paris.
RÉSUMÉ
Summary
»
Dans cet article, plusieurs protéines kinases qui participent au
processus complexe du cycle cellulaire sont décrites. Le rôle clé des
complexes cyclines/CDK dans le contrôle du cycle cellulaire et dans
la prolifération anarchique des cellules cancéreuses est examiné .
Ces données off rent de nouvelles perspectives pour les traitements
anticancéreux. Cependant, les molécules qui bloquent l’activité
des kinases du cycle cellulaire ne sont pas susceptibles de cibler
spécifi quement les cellules tumorales. Les connaissances actuelles
suggèrent que la dérégulation particulière des cyclines/CDK est à
prendre en compte afi n de faciliter le développement des nouveaux
agents antiprolifératifs. Récemment, les inhibiteurs de CDK4/6 se
sont révélés effi caces en combinaison ; ils peuvent être exploités
dans divers types de cancer, en particulier dans le cancer du sein.
Mots-clés : Cycle cellulaire – CDK – Cancer.
In this review, several protein kinases that participate in the
multifaceted process of the cell cycle will be described. The
key role of the cyclins/CDKs complexes in the control of the
cell cycle as well as in the anarchic proliferation of cancer
cells will be defi ned. This data provides new opportunities
for cancer therapy. However, drugs that block the cell cycle
kinases activity are unlikely to selectively target tumor cells.
Evidence suggests that specifi c cyclins/CDKs deregulation has
to be understood to better develop potential anticancer drugs.
Recently, CDK4/6 inhibitors have proven to be eff ective in
combination approaches, which may be exploited in various
cancer types, especially for breast cancers.
Keywords: Cell cycle – CDK – Cancer.
U
ne des clés de voûte de la propagation
des cancers est la prolifération anarchique
des cellules néoplasiques : des divisions cel-
lulaires dérégulées. En eff et, normalement, la division
cellulaire est une succession de processus complexes,
chacun contrôlé par des systèmes de molécules
régulatrices (stimulant ou inhibant la progression
dans le cycle). Il existe donc des points de contrôle
du cycle cellulaire et des signaux permettant l’arrêt
de la division, par exemple en cas de dommage dans
l’ADN. Afi n de passer d’une étape à une autre, des
sérine-thréonine kinases appelées cyclin-dependent
kinases (CDK) sont activées, notamment grâce à leur
interaction transitoire avec leurs protéines parte-
naires : les cyclines.
Dans cet article , les mécanismes principaux de la pro-
gression dans le cycle cellulaire vont être décrits, et
nous insisterons sur le rôle prépondérant des couples
cycline/CDK. Leur ciblage s’avère être une nouvelle
arme thérapeutique pour les patientes ayant un cancer
du sein hormonosensible.
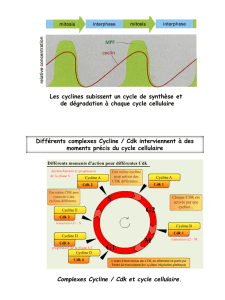
Fonctionnement du cycle cellulaire
Le cycle cellulaire peut être académiquement découpé
en plusieurs phases : G1 (gap or growth phase 1), S (DNA
synthesis), G2 (gap or growth phase 2) et M, M repré-
sentant la mitose proprement dite, avec ses propres
phases (prophase, prométaphase, métaphase, ana-
phase et télophase). Ce découpage correspond à des
événements moléculaires consécutifs, avec des étapes
et des zones de transition entre chaque phase du cycle.
De façon simpliste, ces transitions sont contrôlées par
des kinases activatrices ou inhibitrices, ainsi que par
des destructions protéolytiques de substrats.
Transition G0/G1
Pour commencer “artifi ciellement” la description des
séquences du cycle cellulaire, il est logique de mention-
ner la sortie de la cellule de la période de quiescence (G0)
et sa progression dans la phase G1. À la fi n de cette
phase G1, avant la transition G1-S, la cellule doit passer
un point de contrôle (point de restriction) dépendant
0030_COO 30 13/04/2017 09:29:58

Correspondances en Onco-Théranostic - Vol. VI - n° 1 - janvier-février-mars 2017
31
Le cycle cellulaire : données biologiques et thérapies ciblant les cyclines/CDK
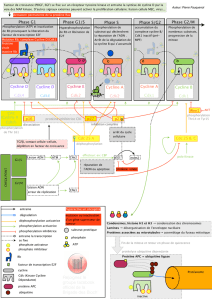
Figure 1. Diff érentes étapes de l’initiation du cycle cellulaire, en réponse à des signaux mitotiques (2).
MAPK
RAF
RAS
PI3K
AKT
Cycline D
Cycline D
Cycline D
Cycline D
Cycline D
CDK4/6 CDK4/6
CDK4/6
CDK4/6
CDK4/6
CDK4/6
Cycline D
Cycline D
Cycline D
Dégradation
par le protéasome
Cytoplasme
GSK3β
(Ub)
n
Récepteur
à l’estrogène
Estrogène
p16
Complexe
inactif
FOXM1
FOXM1
E2F
P
P
P
P
PP
P
Cycline E
CDK2
Rb
E2F
Rb
E2F
Rb
Transcription de gène
dépendant de RE
Passage du point
de contrôle G1
Inactivation de Rb
Expression de gènes associés
à la phase S et à la transition G2/M
Échappement
à la sénescence
Facteur de croissance/récepteur tyrosine kinase
Déplétion
de p16
P
P
P
de certaines protéines appelées “ pocket proteins” , dont
la protéine du rétinoblastome (Rb1). Le passage de ce
point de restriction engage irrémédiablement la cellule
dans sa division. Des signaux promitotiques (voies de
transduction du signal partant des récepteurs membra-
naires à activité tyrosine kinase, par exemple) et des
signaux antiprolifératifs (voie du transforming growth
factor-beta [TGFβ], par exemple) convergent vers les
protéines impliquées dans cette transition G1-S.
Transition G1/S
Sous la pression des signaux promitotiques, les
cyclines D (D1, D2 et D3) [1] s’accumulent et forment
des couples avec les kinases CDK4 ou -6 (fi gure 1) . Il
y a schématiquement 2 voies pour la transition en
phase G1-S impliquant les CDK4/6.
✓
Dans la voie classique, les couples cyclines D/CDK4 ou
-6 phosphorylent partiellement la protéine Rb1 sur diff é-
rents résidus (3) . En eff et, au cours de la phase G1, la pro-
téine Rb1 est hypophosphorylée et couplée au facteur de
transcription E2F pour réprimer son activité (fi xation au
domaine de transactivation) [4] . C’est la phosphorylation
de Rb1 (par CDK4/6) qui libère le facteur de transcription
E2F (fi gure 2, p. 32) . Ce dernier promeut la transcription
d’autres cyclines, à commencer par les cyclines E (5) . Les
cyclines E s’associent avec CDK2 afi n d’initier la synthèse
de l’ADN et, donc, la phase S. Le complexe cycline E/CDK2
phosphoryle à nouveau Rb1, réduisant complètement
son pouvoir inhibiteur sur E2F, et permettant encore plus
de libération d’E2F pour poursuivre la transcription et la
synthèse de cyclines. Il s’agit donc bien d’une autoac-
tivation du cycle cellulaire. La cycline A2, se couplant
aussi avec CDK2, favorise la poursuite de la synthèse
de l’ADN et la transition phase S-G2 (fi gure 3, p. 32) [7] .
Les éléments nécessaires à la synthèse de l’ADN sont
contrôlés essentiellement par 2 kinases : CDC7 et CDK2.
✓
Dans la voie alterne, il semblerait exister une acti-
vation précoce des CDK2 indépendante des étapes
initiales via les couples cyclines D/CDK4/6. Les CDK2
peuvent ainsi se coupler aux cyclines E mais également
aux cyclines D, phosphorylant Rb1. Les mécanismes
conduisant à une activation de CDK2 précoce en cours
de phase G1, indépendamment des CDK4/6, sont mal
connus actuellement.
0031_COO 31 13/04/2017 09:29:58

Correspondances en Onco-Théranostic - Vol. VI - n° 1 - janvier-février-mars 2017
32
dossier thématique
Prolifération
et cycle cellulaire
Figure 2. Processus d’activation de la transcription des gènes du cycle cellulaire par mise en jeu
du couple cycline D/CDK4/6 (qui inhibe Rb et libère le facteur de transcription E2F).
Cycline D
Programme
transcriptionnel
Rb
Rb
E2F
E2F
CDK4/6Ciclib
P
P
CAK
Cdc 25
Gènes du cycle cellulaire : CCNE1, CCNA2, CCNB1, CDK2, CDK1
Gènes de la réplication : MCM2, MCM3, MCM5, MCM7, CDT1, CDC6
Gènes de la mitose : CDC20, PLK1, MAD2L1, CCNB1
ATP
Cycline D
CDK4/6
P
Figure 3. Initiation et progression dans le cycle cellulaire : rôle de Rb et des couples cycline/CDK (6).
M
G1
G2
S
P
P
P
P
P
P
P
P
P
P
Sortie de
quiescence
G0
Cip-Kip
Cip-Kip
Facteurs de croissance
CDK4 et
CDK6 CYCD
RB
RB
RB
RB
CYCE
CYCE
CYCA
CDK2
CDK2
CYCA
CDK1
CYCB
CDK1
INK4
Feedback
positif
Entrée
dans la phase S
Signal activateur
Signal inhibiteur
E2F
E2F
des couples cycline B/CDK1 (appelés aussi CDC2) se
forment et s’accumulent. Les kinases Wee1 et Myt1
(kinases inhibitrices) phosphorylent ce complexe (sur
les 2 résidus Thr-14 et Tyr-15), et le maintiennent ainsi
inactif (fi gure 4) [9] . C’est l’activation de cette grande
quantité de complexes cycline B/CDK1 qui marque l’en-
trée dans la phase M (mitose). Cette activation dépend
de multiples mécanismes, en commençant par l’inhibi-
tion des kinases Wee1 et Myt1 et la déphosphorylation
par les phosphatases CDC25 des 2 résidus (Thr-14 et
Tyr-15), levant ainsi leur action inhibitrice sur le couple
cycline B/CDK1.
Mitose
La mitose est alors un enchaînement d’événements
chromosomiques et du fuseau microtubulaire : conden-
sation des chromosomes, redistribution de la tubuline,
duplication des centrosomes, assemblage du fuseau,
rencontre des microtubules et des kinétochores, et,
enfi n, cytokinèse. L’activité du complexe cycline B/CDK1
intervient dans la condensation des chromosomes et
dans les principaux phénomènes impliqués dans leur
séparation (CDK1 phosphoryle plusieurs dizaines de
substrats) [10] . C’est la protéolyse de la cycline B et,
donc, la chute d’activité de CDK1 qui achève la mitose
en déclenchant la cytokinèse (activation de la sépa-
rase, décondensation des chromosomes et reformation
de l’enveloppe nucléaire). La ségrégation des chro-
mosomes est médiée par le complexe APC/C (ana-
phase-promoting complex/cyclosome) et par certains
cofacteurs comme CDC20. Les cyclines B et A sont des
cibles d’APC/C afi n d’éteindre l’activité de CDK1.
Par ailleurs, d’autres kinases interviennent, dont les
kinases Plk (polo-like kinases) et les kinases aurora. Plk1
phosphoryle la cohésine, qui peut alors être dégradée
(détruite par une caspase), ce qui permet la séparation
des chromatides. Le rôle de l’aurora A, au niveau des
pôles du fuseau mitotique, est de permettre la matu-
ration des centrosomes, leur séparation et la mise en
place du fuseau. L’aurora B est située le long des chro-
mosomes, le plus souvent au niveau du centromère
pour permettre la cytokinèse (fi gure 5) [11] .
Contrôle du cycle cellulaire
Les couples cyclines/CDK s’associent dans une période
de temps relativement courte (liée à une dégradation
rapide via l’ubiquitination des cyclines), ce qui limite la
durée de l’activité kinase . De plus, l’activation et l’inac-
tivation des CDK sont sous le contrôle d’une balance
phosphorylation/déphosphorylation, la phosphoryla-
Transition G2/M
En phase G2, le complexe cycline A/CDK1 est parti-
culièrement actif. En fi n de phase G2, le monomère
CDK1 est présent, et, avec la synthèse importante de
cycline B (parallèle à la dégradation des cyclines A),
0032_COO 32 13/04/2017 09:29:59

Correspondances en Onco-Théranostic - Vol. VI - n° 1 - janvier-février-mars 2017
33
Le cycle cellulaire : données biologiques et thérapies ciblant les cyclines/CDK
Figure 4. Mécanismes de contrôle des cassures de l’ADN impliquant les couples cyclines/CDK.
Lésion de l’ADN Cassure double brin
ATR
CHK2
Wee1
CHK1
PLK1
Aurora A
Cycline
CDC25
Claspine
p53
p21
Myt
ATM
Dégradation
Séquestration CDK
APC/C
14-3-3
tion pouvant être parfois inhibitrice et la déphospho-
rylation (induite par la famille CDC25, par exemple)
activatrice du complexe cycline/CDK.
Il existe 2 familles de protéines contrôlant la progression
dans le cycle : la famille des protéines inhibitrices de
l’action des couples cyclines/CDK (appelée en général
cyclin-dependent kinase inhibitors [CKI] ) , dont font partie
les inhibiteurs de CDK4 (appelés inhibitors of cdK4 [INK4]),
et les inhibiteurs de cyclines/kinases ( CDK interacting
protein/kinase inhibitory protein [CIP/KIP]) [tableau] .
La famille INK4 comporte 4 protéines similaires, p15
INK4B
,
p16
INK4A
, p18
INK4C
et p19
INK4D
, toutes inhibant principa-
lement CDK4 et 6 (et peu les autres CDK) [12] . Pour
l’exemple le plus connu, p16
INK4A
est codé par le gène
CDKN2A (gène suppresseur de tumeur) ; son expres-
sion est induite par de nombreux processus, comme la
sénescence ou la voie du TGFβ. De plus, lorsqu’il existe
une perte de fonction de la protéine Rb1, il existe paral-
lèlement une surexpression de p16
INK4A
.
En ce qui concerne la famille CIP/KIP, il existe 3 pro téines :
p21
CIP1
, p27
KIP1
et p57
KIP2
. Ces protéines sont régulatrices
de toutes les kinases CDK, mais peuvent avoir un eff et
inhibiteur ou activateur, en fonction des complexes
protéiques considérés. Pour simplifier , CIP/ KIP
stabilise le complexe cycline D/CDK4 et permet sa
translocation dans le noyau ; en revanche, il inhibe
CDK2. Le complexe cycline D/CDK4/6 stabilisé favorise
l’expression du complexe cycline E/CDK2, qui lui-même
inhibe (par phosphorylation) CIP/KIP, formant ainsi une
boucle de contrôle négative (figure 3) . La protéine
p27
KIP1
, phosphorylée par le complexe cycline E/CDK2,
est détruite par le protéasome après ubiquitination
(par Skp1/Skp2), ce qui permet l’entrée en phase S (13) .
La synthèse de l’ADN peut aboutir à des dommages
qui sont repérés par des points de contrôle spécifi ques.
Deux kinases sont importantes dans ce contrôle :
ATM (ataxia telangiectasia mutated) et ATR (ATM- and
Rad3-related) . La première est activée par les coupures
double brin, la deuxième est activée par les erreurs
de réplication. ATM et ATR phosphorylent les kinases
Chk2 et Chk1. Chk2 et Chk1 phosphorylent à leur tour
les phosphatases CDC25 et les rendent ainsi inactives
(fi gure 4) . Or, comme nous l’avons souligné ci-dessus,
les CDC25 activent le couple cycline B/CDK1 (en levant
l’inhibition par déphosphorylation sur les résidus Thr-14
et Tyr-15). Les anomalies de la réplication de l’ADN, via
ATM et ATR qui inactivent CDC25, aboutissent donc à
l’inhibition de CDK1 et à l’arrêt du cycle (14) .
De plus, ATM phosphoryle la protéine MDM2, ce qui a
pour eff et d’inhiber son interaction avec le facteur de
transcription p53. Ainsi, p53 est libéré et il est à son tour
phosphorylé par ATM ou par Chk2, ce qui renforce sa
stabilité (les phosphorylations réduisent son ubiquiti-
nation et sa dégradation). La quantité de p53 augmente
et son activité transcriptionnelle augmente en paral-
lèle, induisant la transcription du gène de la protéine
p21
CIP1
. L’expression de p21
CIP1
induit une inhibition de
CDK2 et 1, bloquant ansi le complexe cycline E/CDK2
(fi gure 4) [15] .
Le processus mitotique comporte également un point
de contrôle d’assemblage du fuseau mitotique : SAC
(spindle assembly checkpoint), qui permet de corriger
Tableau. Protéines contrôlant la progression dans le cycle (CKI).
Famille INK4
(inhibiteurs de CDK4)
p15INK4B
p16INK4A
p18INK4C
p19INK4D
Famille CIP/KIP
(inhibiteurs de cyclines/kinases)
p21CIP1
p27KIP1
p57KIP2
0033_COO 33 13/04/2017 09:29:59

Correspondances en Onco-Théranostic - Vol. VI - n° 1 - janvier-février-mars 2017
34
dossier thématique
Prolifération
et cycle cellulaire
Figure 5. Implication des kinases PLK (polo-like kinases) et des kinases Aurora dans le cyle
cellulaire.
P
h
a
s
e
S
Aurora A
Aurora B
Prométaphase Anaphase
Prophase
Télophase
Séparation
des centrosomes
Dissociation
des cohésines le long
des bras des chromosomes
Attachement
des kinétochores Assemblage
du fuseau mitotique
Élongation
du fuseau central
Cytokinèse
Dommages
de l’ADN Entrée
en mitose
Maturation
des centrosomes
Duplication
des centrosomes
Entrée
en phase S PLK3
PLK3
PLK4
PLK4
PLK2
PLK2
PLK1
PLK1
PLK1
Mitose
G2 G1
les anomalies d’attachement des kinétochores aux
microtubules. Il s’agit d’une voie de signalisation impli-
quant aurora B, et d’autres protéines comme MAD1
(mitotic arrest defi cient protein 1) , MPS1 (monopolar
spindle 1) ou BuB (budding uninhibited by benzimidazole) ,
pour inactiver APC/C.
Les kinases du cycle cellulaire
comme cible thérapeutique
Étant donné le rôle prépondérant des kinases du
cycle cellulaire dans les mécanismes de prolifération
des cellules cancéreuses, et la surexpression/déré-
gulation de plusieurs d’entre elles dans les tumeurs,
de nombreux agents à visée anticancéreuse ciblant
ces kinases ont été développés. Ainsi, des inhibiteurs
pan-aurora kinase, des inhibiteurs spécifi ques de l’au-
rora kinase A (MLN8054, MLN8237, ENMD2076), et des
inhibiteurs spécifi ques de l’aurora kinase B (AZD1152,
GSK1070916) ont été testés, mais sans grand succès
pour l’instant. De même, des agents bloquant Plk1
ont été développés (BI2536, BI6727, GSK461364,
HMN241, ON01910), mais l’inhibition de Plk1 a des
conséquences trop pléiotropiques pour être clinique-
ment intéressante (6) .
La voie cycline D −CDK4/6−p16−Rb est également
très souvent dérégulée, offrant donc une cible
thérapeutique de choix. Dans le programme lyon-
nais ProfiLER, proposant une caractérisation molé-
culaire large dans différents modèles tumoraux, nous
avons mis en évidence des altérations de cette voie
chez 16 % des patients (amplifications du gène de
la cycline D1, délétions homozygotes du gène de
p16, ou bien amplifications des gènes codant pour
les CDK) [16] . La cycline D1 est en effet surexprimée
dans de nombreux types tumoraux. Les transloca-
tions t(11;14) des lymphomes du manteau abou-
tissent ainsi à une surexpression de la cycline D1 (17) .
Par ailleurs, une activation anarchique de CDK1 a
été mise en évidence dans de nombreuses tumeurs,
souvent associée à une surexpression de la cycline B1,
et parfois corrélée à un mauvais pronostic. De plus,
CDK2, CDK4 ou CDK6 présentent des activations
aberrantes (le plus souvent à type d’amplification,
les mutations étant rares) dans de nombreux cancers.
Il a donc été développé de nombreux inhibiteurs
des CDK, à commencer par des inhibiteurs pan-CDK.
L’agent le plus connu est le flavopiridol , mais son
activité antitumorale était limitée et sa toxicité non
négligeable. Il est donc apparu logique de cibler
spécifiquement CDK4/6 et, ainsi, d’“épargner” CDK2
afin que les cellules normales puissent poursuivre
des cycles pseudo-normaux.
Trois inhibiteurs de CDK4/6 (ciclib ou CDKi) ont, à ce
jour, un développement clinique avancé. Le palbociclib
(PD-0332991), le ribociclib (LEE011) et l’abémaciclib
(LY2835219) présentent des IC50
assez similaires en ce
qui concerne l’inhibition de CDK4 et CDK6 (2 à 11 nM
pour CDK4, et 10 à 39 nM pour CDK6). En revanche, en
ce qui concerne CDK2, les IC
50
sont assez variables entre
ces 3 molécules (entre 504 nM pour l’abémaciclib et
>10 000 pour le palbociclib), ce qui pourrait expliquer
en partie les diff érences en termes de mode d’adminis-
tration et de toxicités (17) . Dans les études précliniques,
il est apparu nettement que ces traitements n’étaient
effi caces que dans les lignées cellulaires exprimant la
protéine Rb1. Le traitement par CDKi aboutit donc à un
0034_COO 34 13/04/2017 09:30:00
 6
7
6
7
1
/
7
100%