Design, Synthèse, Analyse Structurale et Réactivité de Nouveaux

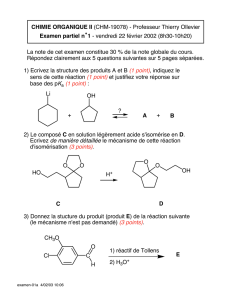
Niculina Daniela BOGDAN
Design, Synthèse, Analyse Structurale et Réactivité
de Nouveaux Cyclophanes
Résumé de thèse
JURY
Pr. Ionel Cătălin POPESCU président Professeur à l’Université "Babeş-Bolyai" Cluj-Napoca
Dr. Chantal ANDRAUD rapporteur Directeur de Recherche au CNRS à l’ENS-Lyon
Dr. Cornelia UNCUŢA rapporteur Directeur de Recherche au C. C. O. «C. D.Neniţescu»,
Bucharest
Pr. Ion GROSU Professeur à l’Université "Babeş-Bolyai" Cluj-Napoca
Pr. Gérard PLÉ Professeur à l’Université de Rouen
Dr. Yvan RAMONDENC Maître de Conférences à l’Université de Rouen
Pr. Patricia MELNYK Professeur à l’Université du Droit et de la Santé de Lille
Pr. Sorin MAGER Professeur à l’Université "Babeş-Bolyai" Cluj-Napoca
Soutenance le 11 juillet 2006

2
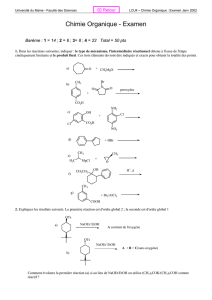
Sommaire
1. Introduction générale
2. Bibliographie: [4.n]cyclophanes
2.1 Introduction
2.2 Généralités
2.2.1 Définition
2.2.2 Nomenclature
2.2.3 Synthèse
2.3 [4.1]Cyclophanes
2.4 [4.2]Cyclophanes
2.4.1 [4.2]Paracyclophanes
2.4.2 [4.2]Métacyclophanes
2.5 [4.3]Cyclophanes
2.5.1 [4.3]Paracyclophanes
2.5.2 [4.3]Métacyclophanes
2.6 [4.4]Cyclophanes
2.6.1 [4.4]Paracyclophanes
2.6.2 [4.4]Métaparacyclophanes
2.6.3 [4.4]Orthoparacyclophanes et [4.4]Orthométacyclophanes
2.6.4 [4.4]Métacyclophanes
2.6.5 Autres [4.4]cyclophanes
2.7 [5.4]-et [6.4]Cyclophanes
3. Nos Résultats :Design, synthèse et analyse structurale des nouveaux
[7.7]cyclophanes et [4.4]cyclophanes comportant des cycles 1,3-dioxaniques
3.1 Introduction
3.2 Rotors moléculaires: design, synthèse, analyse structurale et
complexe d’argent de nouveaux [7.7]cyclophanes
3.2.1 Synthèse et analyse structurale des précurseurs
3.2.2 Synthèse des cyclophanes
3.2.3 Calculs de modélisation moléculaire
3.2.4 Analyse structurale en solution
3.2.5 Aspects structuraux à l’état solide
3.2.6 Conclusion
3.3 Design, synthèse, analyse structurale et réactivité de nouveaux
[4.4]cyclophanes diéthers
3.3.1 Synthèse des intermédiaires
3.3.2 Synthèse des cyclophanes
3.3.3 Analyse structurale en solution
3.3.4 Analyse structurale à l’état solide
3.3.5 Conclusions

3
3.4 Essais de synthèse de cyclophanes stratifiés
3.4.1 Première stratégie
3.4.2 Deuxième stratégie
3.4.3 Conclusion
3.5 Design, synthèse et analyse structurale de nouveaux
[4.4]cyclophanes diesters.
3.5.1 Synthèse des cyclophanes
3.5.2 Analyse structurale en solution
3.5.3 Analyse structurale à l’état solide
3.5.3.1 Composé 39a (monomère)
3.5.3.2 Composé 40a (monomère)
3.5.3.3 Composé 40b (dimère)
3.5.3.4 Composés 41b-d (dimère, trimère et tétramère)
3.5.4 Conclusions
4. Conclusions générales
5. Partie expérimentale
5.1 Indications générales
5.2 Synthèse des [7.7](2,6)pyridinocyclophanes 2a et 5a
5.3 Synthèse du complexe 6a
5.4 Synthèse des composés 9 et 10
5.5 Synthèse du composé 14
5.6 Synthèse du composé 15
5.7 Synthèse du composé 13
5.8 Synthèse des [4.4]cyclophanes 20 – 23 et 25
5.9 Synthèse du composé 26
5.10 Synthèse des monocétones 27 et 28
5.11 Synthèse des dicétones 29 et 30
5.12 Synthèse des composés 32, 34 et 38
5.13 Synthèse du composé 31.
5.14 Synthèse des [4.4]cyclophanes (39a, 40a, 42a) ; [4.4.4.4]cyclophanes
(40b, 41b) ; [4.4.4.4.4.4]cyclophanes (40c, 41c);
[4.4.4.4.4.4.4.4]cyclophane (41d) et [4.4.4.4.4.4.4.4.4.4]cyclophane (41e).
Références
Annexes

4
Introduction générale
Les travaux décrits dans ce mémoire sont focalisés sur l’élaboration, la synthèse,
l’analyse structurale et la réactivité de nouveaux cyclophanes macrocycliques.
Le premier chapitre présente une description générale de la chimie des [4.n]
cyclophanes.
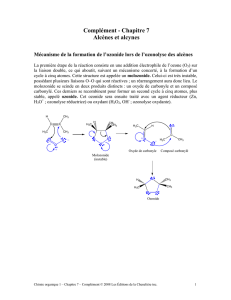
Dans le deuxième chapitre nous nous sommes intéressés à la synthèse, à
l’analyse structurale et à la réactivité d’une série de (2,6)[7.7]pyridinocyclophanes,
d’une série de [4.4]cyclophanes diéthers et d’une série de [4.4]cyclophanes diesters.
N
O
O
O
O
H3C
H3C
CH3
CH2
CH2
CH3
O
O
C
X
C
O
O
n
N
H3C
H3C
O
O
O
O
O
O
H
2
C
H
2
C
CH
3
CH
3
CH
3
CH
3
O
O
X
CH
2
n
R
1
R
2
O
O
O
O
CO
CO
CH
3
CH
3
CH
3
CH
3
O
O
X
CH
2
n
H
3
C
H
3
C
O
O
Les (2,6)[7.7]pyridinocyclophanes présents un comportement des rotors
moléculaires La rotation du cycle pyridinique est plus encombrée dans les dérivés avec
une distance plus grande entre les chaînes (para > méta et 2,6-pyridylen > ortho) et
peut être arrêtée chimiquement par complexation avec AgOTf.
Les [4.4]cyclophanes présentent un intérêt particulier important du fait des
interactions entre les cycles aromatiques (π-stacking). Les éthers sont obtenus
principalement comme monomères et présentent une rotation libre ou encombrée des
cycles aromatiques. Les composés à structures figées sont chiraux (due à la chiralité
planaire du cyclophane). Par réaction d’hydrolyse, des mono et dicétones
macrocycliques ont été préparées. Au cours de ces synthèses, nous avons observé une
stabilité inhabituelle des cycles 1,3-dioxaniques à cette réaction d’hydrolyse.

5
R
R
1
O
O
O
O
O
O
R
R
1
O
O
O
O O
R
R
1
O
OO
O
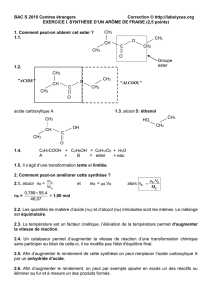
Les [4.4]cyclophanes diesters sont intéressant comme molécules hôtes car ils
présentent des cavités de tailles (n=1-5) et géométries différentes. D’importantes
interactions π- π stacking, CH—π et CO—π ont été mise en évidence tant à l’état
solide qu’en solution.
3. Nos résultats: Design, synthèse et analyse structurale des nouveaux
[7.7]cyclophanes et [4.4]cyclophanes comportant des cycles 1,3-dioxaniques
3.1 Introduction
Les cyclophanes sont des cibles synthétiques attractives pour leurs propriétés
chimiques, physico-chimiques et biologiques particulières.1,2,3,4. Ce sont des exemples
typiques de dérivés comportant la chiralité planaire.5
Les [2.2]paracyclophanes chiraux sont de plus en plus étudiés et utilisés comme
ligands et auxiliaires chiraux dans des synthèses asymétriques.6,7,8,9,10,11,12
Ces dernières années, un grand intérêt a été montré pour la synthèse de
cyclophanes macrocycliques ayant différentes cavités. Ceux-ci peuvent fonctionner
comme molécules «hôtes».13,14,15,16,17,18,19 Beaucoup de cyclophanes ont été étudiés
comme rotors moléculaires.20,21,22,23
Les interactions non covalentes sont présentes et de grande importance dans
beaucoup de domaines de la chimie, de la biologie et des sciences des matériaux.24 En
particulier, les interactions π-π stacking sont fondamentales dans les processus
d’organisation supramoléculaire et de reconnaissance moléculaire. 25
Un grand nombre de propriétés des cyclophanes à l’état solide ou en solution sont
dues à des interactions entre les cycles aromatiques. Beaucoup d’études théoriques et
expérimentales portent sur la géométrie et l’importance des interactions π–
π.26,27,28,29,30,31,32
Les calculs réalisés sur le dimère du benzène ont mis en évidence cinq
arrangements différents (Figure 1): face-to-face (VI), offset-stacked (VII), edge-to-face
(en T, VIII), edge-titled-T (XI), et face-titled-T (X).33
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
1
/
56
100%