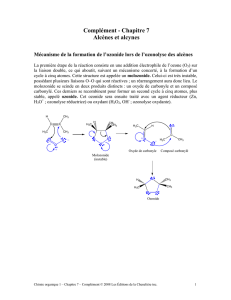
Induction asymétrique: profil énergétique

INTRODUCTION AUX REACTIONSINTRODUCTION AUX REACTIONS
STEREOSELECTIVES.STEREOSELECTIVES.
Aspects générauxAspects généraux
..
Introduction aux réactions stéréosélectives
Aspects générauxAspects généraux
..
Maîtrise Option Chimie Organique
2005
"L’univers est dissymétrique“
Louis Pasteur (1893)
Contact: Pr. Pierre van de Weghe, UMR 6226 Sciences Chimiques de Rennes
Equipe Produits Naturels, Synthèses, Chimie Médicinale (bât 5, rdc)
pierre.van-de-weghe@univ-rennes1.fr
http://blogperso.univ-rennes1.fr/pierre.van-de-weghe/index.php/
tél : 02 23 23 38 03

Introduction aux réactions stéréosélectives
Pré requis
relations de stéréochimie (diastéréiosomérie, énantiomérie)
détermination des configurations absolues des centres stéréogènes (Rou S)
détermination des excès diastéréoisomériques et énantiomériques
analyse conformationnelle (systèmes acycliques et cycliques)
réactivité
de
base
des
grandes
fonctions
de
la
chimie
organique
réactivité
de
base
des
grandes
fonctions
de
la
chimie
organique
Ouvrages conseillés pour étoffer vos connaissances liées à ce cours
C. Rabiller Stéréochimie et chiralité en chimie organique, DeBoeck université.
Carey – Sundberg Tomes 1 & 2
M. Nogradi Stereoselective Synthesis, VCH.
E.L. Eliel, S.H. Wilen, M.P. Doyle Basic Organic Stereochemistry, Wiley Interscience.
J. Seyden-Penne Synthèse et catalyse asymétriques, CNRS Editions.

Introduction aux réactions stéréosélectives
Généralités
Chiralité en chimie
molécules chirales: composés différents de leur image dans un miroir.
nomenclature des énantiomères: application de la règle de Cahn-Ingold-Prelog.
H
3
CH
CO
2
H
OH
(
S
)
-
a
c
i
d
e
l
a
c
t
i
q
u
e
1
2
34
C
H
3
C
HH
CH
3
a
l
l
è
n
e
(
R
)
CO
2
H
OH
HO CO
2
H
(
R
,
R
)
-
a
c
i
d
e
t
a
r
t
r
i
q
u
e
(
S
)
-
a
c
i
d
e
l
a
c
t
i
q
u
e
a
l
l
è
n
e
(
R
)
(
R
,
R
)
-
a
c
i
d
e
t
a
r
t
r
i
q
u
e
QUIZZ: parmi les composés suivants, indiquer ceux qui présentent une isomérie optique (composés chiraux).
NEt
CH
3
Ph
butan-2-ol Ph PEt
CH
3
HO
2
C
H
CH
3
H
NO
2
NO
2
NO
2
HO
2
C
CO
2
H
NO
2
CH
3
CH
3
CO
2
H

Introduction aux réactions stéréosélectives
excès
énantiomérique
–
excès
diastéréoisomérique
.
il y a stéréoisomérie quand deux composés ont la même formule plane (énantiomérie et diastéréoisomérie).
Pour un substrat possédant deux atomes de carbone chiraux:
R,R
S,S
R,S
S,R
diastéréoisomérie
énantiomérie
OH
O
OH
OH
thréose
OH
O
OH
OH
érythrose
OH
O
OH
OH OH
O
OH
OH
excès
énantiomérique
–
excès
diastéréoisomérique
.
e.e. = [R] - [S]
[R] + [S]x 100 e.d. = [R,R] - [S,R]x 100
[R,R]+ [S,R]
prochiralité:
B H
A
H
B H
A
D
B D
A
H
carbone
prochiral
O
H
3
C
Ph
H
3
C
Ph OH
H
H
3
C
Ph H
OH
faces prochirales
ré
si

Introduction aux réactions stéréosélectives
QUIZZ: relation de topicité: indiquer la relation existant entre les groupes soulignés (énantiotopes etc…).
O
H
3
C CH
3
H
3
C CH
3
O
O
H
3
C
H
3
C
O
H
3
C CH
3
H
3
C CH
3
QUIZZ: on considère le substrat ci-dessous. Indiquer la configuration absolue des centres chiraux. La fonction
aldéhyde subit une réaction de réduction, le composé obtenu est-il chiral? Justifiez votre réponse.
OH OHO
OH OH
H
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
1
/
92
100%