25 Mucoviscidose

UE Génétique médicale Typeur : HUNAULT Marine
Cours n° 25 – Dr Demeer - 27.03.2014 Correcteur : JAAFAR Ali
1
LA MUCOVISCIDOSE
C’est une pathologie très fréquente, la fréquence de la mutation CFTR est de l’ordre de 1/25 à 1/30.
Il existe un risque de 25% de transmission autosomique récessive avec 2 parents hétérozygotes
porteurs.
I. Gène CFTR et ses mutations
A. Généralités
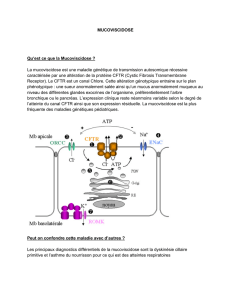
- La mucoviscidose est due à une mutation du gène CFTR (cyystic fibrosis transmembran
receptor) qui code pour un canal chlore, ce qui explique la majorité des signes associés à la
mucoviscidose : anomalies des échanges d’ions chlore et sodium : mucus qui tapisse les
parois s’épaissi les bactéries/poussières ne sont plus éliminé.
- Elle a tout d’abord été décrite comme une fibrose kystique pancréatique.
- On décrit beaucoup de mutations au niveau de ce gène (1800 à ce jour). La plus fréquente
est la mutation F 508 (= délétion).
- La mutation et son effet dépendent de l’origine ethnique +++ (demander lors de
l’interrogatoire).
- On a classé les mutations en 5 classes différentes selon leur impact protéique.
o Mutations sévères : I, II, III et mutations peu sévères : IV, V.
o Classe I : défaut de production de la protéine.
o Classe II : défaut de maturation de la protéine.
o Classe III : anomalie de régulation.
o Classe IV : anomalie de conduction du canal chlore.
o Classe V : réduction du taux d’ARN messager ou de la protéine.
B. Les anomalies biochimiques
- Sécrétion anormales des muqueuses.
- Altération des mouvements ioniques, en partie du chlorure au niveau de la membrane
apicale.
C. Conséquences du blocage de la sécrétion du chlorure et de l’eau
- Epaississement du mucus.
- Modification de la composition ionique de la sueur (test de la sueur).
- Anomalie du potentiel trans-épithélial (patch-clamp in vitro).
II. Signes cliniques
- Schéma
o Coup de chaleur.
o Sinusite, polypose nasale.
o Toux, RGO.
o BPCO, DDB, surinfection bronchique.
o Cirrhose, HTP.

2
o Cholestase.
o Péritonite méconiale.
o Invagination.
o Iléus méconial.
o Stérilité masculine, retard pubertaire.
o Prolapsus rectal.
o Diarrhée chronique.
o Volvulus.
o Mal digestion, pancréatite, diabète.
o HTAP.
III. Diagnostic clinique
- Variabilité d’expression +++
- Atteinte pulmonaire et digestive.
- Clinique +++
A. Forme classique
- Atteinte pulmonaire : BPCO.
o Obstruction par mucus épais, abondant.
o Toux chronique.
o Altération de la fonction respiratoire.
o Infection pulmonaire (S. aureus, H. influenzae puis Ps. Aeruginosa).
o Evolution : bronchestasies (DDB), cœur pulmonaire chronique.
B. Atteinte du tractus gastro-intestinal :
- Iléus méconial néo-natal (15%).
- Obstruction iléale.
- Prolapsus rectal.
C. Insuffisance pancréatique exocrine (85% des NN)
- Diarrhées fétides, graisseuses.
- Hypotrophie, ballonnement abdominal.
- Carence protidique, vit. liposolubles.
D. Autres signes
- Retard staturo-pondéral.
- Dysfonctionnement du pancréas endocrine (diabète par fibrose pancréatique).
- Cirrhose (biliaire), ictère rétentionnel du foie.
- Problèmes génito-urinaires (stérilité masculine).
- Polypose naso-sinusienne.
- Coup de chaleur hyponatrémique (perte accrue de sel).
E. Formes frontières : CFTR-RD (related disorder)
- Stérilité par absence bilatérale des canaux déférents (ABCD), azoospermie sécrétoire.
o + Fréquente stérilité masculine.
o 50% agénésies : 1 mutation et + (30% : 1 mutation + 1 variant).
o Conseil génétique, possibilité FIV/ICSI.

3
- Pancréatites chroniques.
- Dilatation des bronches (DDB).
- Aspergillose broncho-pulmonaire atypique.
IV. Diagnostic para-clinique
- Examens complémentaires :
o NN : trypsine immuno-réactive.
o Enfant : test de la sueur : concentration en ion chlorure et sodium.
Positif quand >60mEq/L à plusieurs reprises (on ne fait pas le test chez les
prématurés).
Positif dans 98% des cas, spécifique de la pathologie.
Aucune relation entre les valeurs du test et la gravité de la maladie.
Impossibilité d’identifier les transmetteurs.
Attention : faux négatifs et surtout faux positifs.
- RX thorax : distension thoracique, impactions mucoïdes, emphysèmes.
- Bilan sanguin.
- Stéatorrhée (selles grasses).
- Prise en charge multidisciplinaire en milieu spécialisé.
- Moléculaire (spécifique) :
o Recherche mutations fréquentes : kit, 30ain mutations, génotype : 60% cas
37 exons, 180 kb.
Résultat en 8 jours.
o Séquençage du gène.
o Recherche de grands réarrangements.
o Taux de couverture : 95-98%.
o Reste 1 à 2% pas de génotype complet (diagnostic para-clinique impossible).
V. Diagnostic positif
- Critères de consensus :
o Association chez un patient d’un ou plusieurs traits phénotypiques de la maladie ou
existence d’un apparenté atteint ou existence d’un test de dépistage néonatal
positif.
Et
o présence d’un est de la sueur positif en 2 occasions ou présence d’une
mutation causale en double exemplaire.
VI. Dépistage néo-natal
- > 10 ans en France.
- 2 étapes :
o TIR à J3 (sensibilité).
o Recherche mutations fréquentes CFTR.
- But : diagnostic précoce.
o Pour prise en charge précoce au sein des CRCM (Centre de Ressources et de
Compétences sur la Mucoviscidose).
o Consultation génétique.

4
o Schéma
VII. Prise en charge
- Equipe pluridisciplinaire, centre spécialisé.
- Pas de traitement curatif.
- Pulmonaire :
o Kinésithérapie, aérosols.
o ATB thérapie anti-pyocyanique discontinue.
o Vaccinations : grippe, pneumocoque.
- Alimentation : régime hypercalorique, hyper-protidique, vit liposolubles, NaCl.
- Pancréatique : extraits pancréatiques.
- Formes avancées : transplantation pulmonaire, voire hépatique.
VIII. Conseil génétique
- Maladie AR : risque de ¼ de récurrence pour l’enfant suivant lorsqu’un couple a eu un 1er
enfant atteint.
- Un enfant diagnostiqué : dépistage familial +++.
o Pour la fratrie d’un parent d’un enfant atteint :
Risque d’être également porteur : ½.
Risque que le conjoint soit porteur : 1/30.
Risque de transmission simultanée : ¼.
Risque d’avoir un enfant atteint = ½ x 1/30 x ¼ = 1/240.
- Possibilité de DPN, DPI.
IX. Diagnostic au cours de la grossesse
- Signes d’appel : intestin hyper-échogène (2T).
o Etude du gène CFTR chez le couple.
Si hétérozygotie/parents : étude gène CFTR fœtal.
o Associé dans 3 à 9% des cas à diagnostic in utero.
- Couple à risque : ¼.
o Etude gène CFTR fœtal.
Choriocentèse (11 SA)/ amniocentèse (16 SA).
- Possibilité demande IMG :
o Conseil génétique : connaissances, évolution clinique, accompagnement/décision,
risque 1/4 par grossesse.
1
/
4
100%