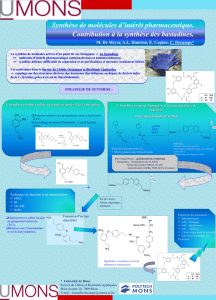
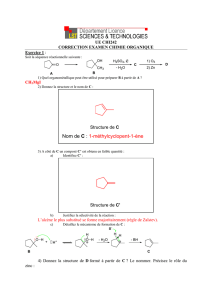
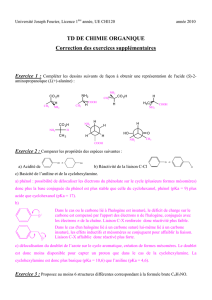
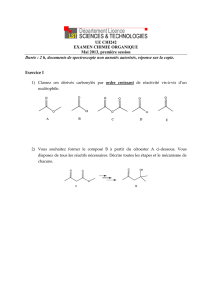
Description des principales fonctions EC1 : Chimie organique PACES 2016-2017

Description des principales fonctions
PACES 2016-2017
Professeur Pascal Marchand
IICiMed, Laboratoire de Chimie Thérapeutique
Faculté de Pharmacie de Nantes
UE1 : Atome - Biomolécules – Génome - Bioénergétique – Métabolisme
EC1 : Chimie organique

• Remplacement d’un ou de plusieurs Hpar un ou plusieurs Xsur un carbone
• Formule générale :
RX
≡
CX
1. Dérivés halogénés non aromatiques
•Acycliques : Exemples :
CH3CH
CH3
I
2-iodopropane
CH3C
CH3
CH2
CH3
Br
1-bromo-2,2-diméthylpropane
Nomenclature systématique : halogène considéré comme un substituant de l’alcane
= halogénoalcanes
Nomenclature plus ancienne : halogénures d’alkyle
CH3Cl Chlorométhane (chlorure de méthyle)
CHCl3Trichlorométhane (trichlorure de méthyle)
Chloroforme
CBrCl3Bromotrichlorométhane (bromure de trichlorométhyle)
Les dérivés halogénésLes dérivés halogénés
I- Définition et nomenclature

• Dérivés aromatiques « à halogènes séparés » : halogénures benzyliques
Exemple :
CH2Br
(bromure de benzyle)
(bromométhyl)benzène
II- Structure
CX
-I +
CX
δ
δ-
Polarisation de la liaison
ELCF > ELCCl > ELCBr > ELCI
Electronégativité : les halogènes sont plus électronégatifs que le C
Taille de l’halogène croissante Electronégativité décroissante

R CH2
X
R
CH
X
R'
R
C
X
R'
R"
Primaire Secondaire Tertiaire
Taille de la liaison :
Exemple :
Existence de 3 classes de monohalogénoalcanes :
CCl
1,76 Å

III- Propriétés chimiques
1. Réactivité générale
1.1 Polarisabilité de la liaison
+
CX
δ
δ-
CCl
δδ
Exemples :
CH2CH CH2Cl
δδ
CH2Cl
δδ
La polarisabilité augmente en fonction :
De la diminution de l’énergie de liaison
De l’augmentation de la taille de X
F << Cl < Br < I
Dérivés R-F sont presque inertes chimiquement
Les dérivés chlorés, bromés et iodés représentent une
source potentielle de carbocations
= ce sont des composés électrophiles (sites pauvres en électrons)
Réactivité croissante
La liaison C-F est très stable Pas de SN ni de β-élimination
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
1
/
38
100%