resume

Résumé du protocole PRODIGE 11 version 1.1 du 12/ 11/ 2009
RESUME
Titre ESSAI RANDOMISE PHASE III SORAFENIB - PRAVASTATINE VERSUS SORAFENIB
SEUL POUR LE TRAITEMENT PALLIATIF DU
CARCINOME HEPATOCELLULAIRE CHILD-PUGH A
PRODIGE 11 - ESSAI ANGH-FFCD 0803
Promoteur Centre Hospitalier Universitaire (CHU) de Dijon - 1 Bd Jeanne d'Arc - BP77908 - 21079
Dijon Cedex
Coordonnateurs Jean-Louis Jouve et Jacques Denis
Pathologie Carcinome hépatocellulaire (CHC) non accessible à un traitement à visée curative
Objectifs Objectif principal : évaluer l’influence de l’association pravastatine – sorafenib
versus sorafenib seul sur la survie globale de patients atteints d’un carcinome
hépatocellulaire inaccessible à un traitement à visée curative et développé sur
cirrhose classée Child-pugh A.
Objectifs secondaires : évaluer l’effet de ce traitement sur la survie sans
progression, le temps jusqu’ à progression, le temps jusqu’à échec du
traitement et la qualité de vie (QLQ-C30 et FACT HEP)
Etude méthodologique : Evaluer par un essai randomisé l’impact de l’ajout d’un
monitoring 100% sur site au cours de l’essai comparé à un monitoring centralisé
par data management sur le taux de dossier patients avec une erreur pour au moins
une des variables majeures recueillies dans l’essai.
Méthodologie Etude de Phase III randomisée, contrôlée, ouverte, multicentrique
Critères
d’inclusion
Carcinome hépatocellulaire diagnostiqué :
- soit par un examen histologique
- soit si la preuve histologique ne peut être obtenue (ascite, troubles de la
coagulation) le diagnostic pourra être porté en cas de cirrhose selon les
critères de l’EASL/AASLD 2005, par la mise en évidence d’une lésion focale
hépatique de plus de 10 mm
- sur deux techniques d’imagerie dynamique (scanner hélicoïdal, IRM,
échographie avec produit de contraste) pour les tumeurs de moins de 2 cm. La
lésion doit avoir pour caractéristiques une hypervascularisation au temps
artériel et un lavage au temps portal tardif
- sur une technique d’imagerie dynamique (scanner hélicoïdal, IRM,
échographie avec produit de contraste) pour les tumeurs de plus de 2 cm avec
hypervascularisation au temps artériel et un lavage au temps portal tardif
Malade non éligible pour un traitement à visée curative (transplantation, résection,
destruction percutanée) ou pour une chimio-embolisation ou ayant un CHC encore
en évolution après échec d’un traitement spécifique
Score pronostique du CLIP 0 à 4
Classe A de Child-Pugh
Transaminases ≤ 5 N et Créatininémie ≤ 1,5 N
OMS : 0, 1 ou 2
Age de plus de 18 ans
Survie prévisible > 12 semaines
Possibilité de suivi régulier
Consentement écrit après information
Critères de non
inclusion
Maladie extra-hépatique engageant le pronostic vital à court ou moyen terme
Autre pathologie cancéreuse en évolution à l’exception du cancer du col utérin in
situ, d’une tumeur superficielle de vessie et d’un carcinome baso-cellulaire traité.
Tout autre cancer traité à visée curative depuis plus de 3 ans peut être inclus dans
l’étude
Insuffisance cardiaque (≥ classe 2 NYHA), HTA ou arythmie non contrôlées,
infarctus du myocarde de moins de 6 mois
Hémorragie digestive de moins de 1 mois
Malade recevant ou ayant déjà reçu un traitement par statines ou par sorafenib
Grossesse et allaitement. Les femmes en âge de procréer devront utiliser une
méthode contraceptive efficace tout au long de l’essai et 3 mois après l’arrêt du
traitement.

Résumé du protocole PRODIGE 11 version 1.1 du 12/ 11/ 2009
2
Bilan Pré-
Thérapeutique
1) Clinique (< 2 semaines avant inclusion) : identification, histoire et étiologie de la
cirrhose, poids, examen clinique, état général (échelle OMS), questionnaires de qualité de
vie QLQ-C30 et FACT HEP
2) Biologie (< 2 semaines avant inclusion) : NFS, plaquettes, TP, albuminémie,
bilirubinémie totale, ASAT et ALAT, phosphatases alcalines, créatininémie, natrémie,
kaliémie, FP, cholestérol total, triglycérides, CPK. Antigène HBs et anticorps anti-VHC (<
3 mois avant inclusion sauf si infection antérieurement connue).
3) Examens morphologiques (< 1 mois avant inclusion) : TDM thoraco-abdominale
ou IRM abdominale et TDM thoracique. Détermination de la masse tumorale selon les
critères d’évaluation du RECIST (version 1.1) et recherche d’une thrombose portale
(tronculaire ou lobaire)
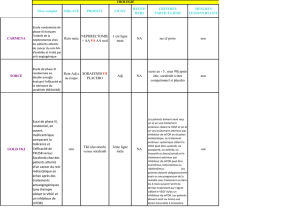
Traitement - Bras A sorafenib-pravastatine : sorafenib 800 mg/j en 2 prises orales –
pravastatine 40 mg/j en une prise per os
- Bras B sorafenib : 800 mg /j en 2 prises orales
Les patients recevront ce traitement jusqu’au décès ou jusqu'à l'arrêt du traitement motivé
par :
- un effet indésirable
- le refus du patient à poursuivre
- une dégradation de l’état général OMS ≥ 3
Une progression radiologique seule, avec cependant description par le patient d’une
amélioration de sa qualité de vie, n’est pas un critère d’arrêt du traitement protocolaire. La
décision est laissée à l’appréciation du patient et de l’investigateur.
Suivi des patients Toutes les 4 semaines, consultation avec le résultat des examens biologiques suivants :
NFS, plaquettes, TP, albuminémie, bilirubinémie totale, ASAT et ALAT, phosphatases
alcalines, créatininémie, natrémie, kaliémie, FP, cholestérol total, triglycérides et CPK.
A cette occasion les questionnaires de qualité de vie QLQ C-30 et FACT HEP seront
remplis par le patient.
Toutes les 12 semaines imagerie identique à celle réalisée lors du bilan initial.
Nombres de
patients
Etude clinique : sorafenib-pravastatine versus sorafenib seul.
L’hypothèse principale est de faire passer la médiane de survie globale de 10 mois dans le
bras sorafenib seul à 13,5 mois dans le bras sorafenib-pravastatine.
Avec un risque =0,05 (test bilatéral) et =0,20, un rythme d’inclusion de 15 patients par
mois pendant 30 mois et 5% de perdu de vue, il sera nécessaire de recruter 474 patients
pour observer les 360 décès nécessaires.
Une analyse intermédiaire est planifiée à la moitié des décès observés pour s’assurer de
l’innocuité et de l’efficacité de l’association. Méthode utilisée : O’Brien – Fleming.
Description succincte de l’étude méthodologique :
Etude méthodologique randomisée de supériorité, en double aveugle dont l’objectif
principal est d’évaluer l’impact de l’ajout d’un monitoring 100% sur site au cours de l’essai
comparé à un monitoring centralisé par data management sur le taux de dossier patients
avec une erreur pour au moins une des variables majeures recueillies dans l’essai : critère de
stratification, critères d’éligibilité, critères de jugements (décès, progression, réponse, date
de décès, dates de dernières nouvelles, date de progression et date de meilleure réponse).
Durée estimée de
l’étude
46 mois
Période estimée
d’inclusion
32 mois
Date de début Novembre 2009
Date de Fin Septembre 2013
1
/
2
100%