Complexe de Pick - STA HealthCare Communications

La revue canadienne de la maladie d’Alzheimer • Mars 2001 • 5
Le terme « maladie de Pick »
désigne des cas cliniques bien
définis de dégénérescence frontale et
temporale progressive, telle que décrite
par Arnold Pick.1Ce même terme
désigne une entité pathologique définie,
du point de vue histologique, par la
présence d’inclusions globulaires argy-
rophiles (corps de Pick) et des neurones
achromatiques gonflés (cellules de
Niemann-Pick). Lorsque Pick a décrit
pour la première fois le cas d’un patient
atteint d’une aphasie progressive accom-
pagnée d’un trouble du comportement
et, plus tard, d’autres cas de démence par
atrophie du lobe frontal avec aphasie, il
a déterminé seulement les caractéris-
tiques anatomiques, définissant plus tard
les caractéristiques histologiques.
Par la suite, dans des cas de maladie
de Pick d’importance clinique, associée
à une atrophie frontale et temporale, les
autopsies ont révélé l’absence des ca-
ractéristiques histologiques types à l’au-
topsie. Après avoir passé en revue un
grand nombre de leurs propres cas,
Constantinidis et ses collègues2ont
établi la classification suivante de la
maladie de Pick : a) maladie de Pick
accompagnée de corps de Pick; b) ma-
ladie de Pick accompagnée seulement
de neurones gonflés; c) maladie de Pick
accompagnée seulement d’une gliose et
d’une perte de neurones. De l’avis de
ces chercheurs, « malgré les dissem-
blances entre ces formes de la maladie
de Pick, compte tenu de l’absence de
connaissances suffisantes au sujet de la
pathogenèse, il [était] sage [...] de
préserver le caractère unique de l’entité
de Pick ». De nombreux autres articles
publiés plus tard sur la maladie de Pick
étaient fondés sur les conclusions de
l’autopsie, et les caractéristiques cli-
niques et variables étaient obtenues de
manière rétrospective. Cette situation a
alimenté l’idée que la maladie de Pick
est difficile à diagnostiquer in vivo.
Démence fronto-temporale
Grâce à la neuroimagerie, la présence
d’une atrophie fronto-temporale a pu
être démontrée de plus en plus souvent
chez des sujets vivants. Par contre, au
lieu d’axer de nouveau le diagnostic de
maladie de Pick vers le tableau cli-
nique, des auteurs d’études plus
récentes ont conçu de nouvelles appel-
lations, par exemple, « démence de
type lobe frontal » ou « démence
frontale ». Les groupes ayant décrit
l’entité « démence de type lobe
frontal » sont ensuite arrivés avec un
autre néologisme, soit la dégénéres-
cence fronto-temporale.3Tous ces
chercheurs reconnaissaient par ailleurs
que le syndrome clinique est iden-
tique : les patients présentent des corps
de Pick ou seulement une perte de neu-
rones et une gliose. Ils évaluaient à
20 % l’incidence des démences
Complexe de Pick : éliminer la
confusion au sujet de la maladie de
Pick et de la démence par atrophie
fronto-temporale
Les progrès des techniques diagnostiques se sont traduits par des taux croissants de diagnostic
d’affections comme l’atrophie frontale et temporale. Malgré tout, il reste difficile de reconnaître
et de distinguer les syndromes cliniques de ces affections et d’autres affections connexes.
Cependant, un des moyens de mieux les comprendre serait de définir une nouvelle terminologie
pour classer et pour relier ces troubles.
par Andrew Kertesz, M.D., FRCPC
Le Dr Kertesz est professeur au
Département de sciences
neurologiques cliniques de la
University of Western Ontario et
directeur du Cognitive Neurology
and Alzheimer Research Center au
St. Joseph’s HealthCare – St. Joseph’s
Hospital de London (Ontario).

dégénératives. Une importance pré-
pondérante était accordée aux troubles
du comportement et, aujourd’hui, le
terme « dégénérescence fronto-tempo-
rale » est utilisé pour désigner le syn-
drome de démence avec apathie et
désinhibition ainsi que comme sy-
nonyme de la maladie de Pick.
Aphasie progressive primaire
Le même phénomène de création d’un
nouveau terme pour désigner la maladie
de Pick est survenu lorsque des
chercheurs ont classé l’aphasie progres-
sive primaire comme une entité dis-
tincte.4Il faut savoir toutefois que dans
plusieurs cas subséquents (et antérieurs)
d’aphasie progressive primaire, on avait
mentionné la présence de corps de Pick.
Dans d’autres cas encore, le tableau his-
tologique incluait une gliose, une perte
de neurones et une spongiose des
couches II et III du cortex, caractéris-
tiques identiques à celles décrites dans la
démence de type lobe frontal, ainsi que
des lésions sous-corticales avec une
achromasie neuronale semblable à celle
décrite dans la dégénérescence corti-
co-basale.5La forme d’aphasie progres-
sive primaire avec discours réduit
entraîne souvent un mutisme qui ne peut
être distingué de celui de la démence
frontale. D’autres aspects du langage
sont ensuite touchés, particulièrement
des changements du comportement évo-
quant un déficit du lobe frontal. De plus,
on observe parfois des complications
extrapyramidales et une sclérose latérale
amyotrophique (SLA). Il existe aussi
une forme intéressante, caractérisée par
la préservation de la fluidité du discours
et de la syntaxe, mais avec une perte de
la compréhension sémantique, forme
dite « démence sémantique ».6
Syndrome de dégénérescence
cortico-basale
Plusieurs cas de maladie de Pick ont été
décrits chez des personnes qui présen-
taient des caractéristiques extrapyrami-
dales. Les chercheurs ont alors reconnu
que des altérations sous-corticales
surviennent dans la maladie de Pick,
même en l’absence de symptômes
extrapyramidaux. Lorsque Rebeiz et ses
collègues7ont décrit la dégénérescence
du cortex, du gyrus dentatus et du locus
niger, ils ont reconnu que cette patholo-
gie était semblable à la maladie de Pick.
Plus tard, le syndrome apraxique extra-
pyramidal, avec paralysie du regard et
« main extra-terrestre » (alien hand syn-
drome), a été désigné par un néologis-
me : la dégénérescence cortico- basale8
ou dégénérescence ganglionnaire corti-
6• La revue canadienne de la maladie d’Alzheimer • Mars 2001
Glossaire
Démence fronto-temporale. Maladie de Pick d’importance clinique ou complexe de Pick.
Cette appellation décrit également un tableau clinique incluant l’apathie et la désinhibi-
tion ou une pathologie sans corps de Pick. Elle a remplacé le terme « démence de type
lobe frontal ». En plus des formes avec apathie et désinhibition, on distingue aussi la forme
avec stéréotype compulsif dans le tableau clinique initial.
Corps de Pick. Inclusions compactes, rondes, argyrophiles, dans le gyrus dentatus et le
néocortex. Certains auteurs sont d’avis que ces inclusions définissent la maladie de Pick,
mais on observe divers types d’inclusions dans le complexe de Pick et, parfois, l’absence
d’inclusion, alors que le syndrome clinique est le même.
Cellules de Niemann-Pick. Neurones gonflés. Les cellules de Pick sont une caractéristique
de toutes les composantes du complexe de Pick, et ces neurones gonflés ont d’abord été défi-
nis dans la maladie de Pick, mais plus tard, comme un signe cardinal de la dégénérescence
cortico-basale. On observe également dans toutes les composantes du complexe de Pick une
spongiose des couches superficielles du cortex, une gliose et une perte de neurones.
Aphasie progressive primaire. Syndrome initial faisant également partie du complexe de
Pick ou de la maladie de Pick d’importance clinique. Plusieurs pathologies sont associées,
tout comme dans la démence fronto-temporale. On observe chez le patient une aphasie
d’évolution lente avant l’apparition de tout autre symptôme. Le discours, d’abord
amnésique, devient de plus en plus réduit.
Démence sémantique. L’aphasie sémantique, c’est-à-dire une forme d’aphasie sensorielle
transcorticale dans laquelle le discours n’est pas réduit et qui est caractérisée par une difficulté
à comprendre et à nommer, mais avec la préservation de la fluidité du discours et de la syn-
taxe. L’incapacité de donner un sens touche aussi les stimuli visuels.
Dégénérescence cortico-basale. Forme extrapyramidale de la maladie de Pick. Définie, du
point de vue clinique, par des symptômes extrapyramidaux unilatéraux, une apraxie et le
syndrome de la « main extra-terrestre », chez de nombreux patients, on observe des carac-
téristiques de démence fronto-temporale et d’aphasie progressive primaire et des syndromes
cognitifs d’emblée présents. Par conséquent, l’entité pathologique de dégénérescence corti-
co-basale ainsi que le syndrome clinique de dégénérescence cortico-basale font partie du
complexe de Pick.
Paralysie pseudo-bulbaire progressive. Forme de paralysie reliée à la dégénérescence
cortico-basale des points de vue pathologique, génétique et clinique. Démontrée par des
recherches récentes, cette relation est importante, mais controversée.
Démence reliée à une SLA. D’abord décrite comme une entité distincte, caractérisée par
la présence d’inclusions particulières, tau-négatives et synucléine-négatives et ubiquitine-
positives, forme de démence ayant permis de constater qu’il y avait un changement impor-
tant entre ces cas et d’autres composantes du complexe de Pick. Ce ne sont pas tous les
patients qui présentent les inclusions caractéristiques, et on observe parfois ces inclusions
en l’absence d’une SLA.
FTDP-17. Démence fronto-temporale et parkinsonisme reliés au chromosome 17. Dans la
plupart de ces familles, les sujets présentent des mutations de la protéine tau, mais d’autres
n’en présentent pas ou, encore, sont atteints d’une pathologie tau-négative.
Complexe de Pick. Maladie de Pick d’importance clinique englobant tous les syndromes de
démence fronto-temporale, d’aphasie progressive primaire, de dégénérescence cortico-basale
et toutes leurs variétés pathologiques, y compris la maladie de Pick, la dégénérescence corti-
co-basale, la démence sans histologie distincte et la démence reliée à une SLA.

co-basale. La plupart des patients
atteints d’une dégénérescence cortico-
basale manifestent un trouble du langage
qui ressemble à l’aphasie progressive
primaire et à la démence fronto-
temporale, les deux syndromes se
chevauchant de façon importante. On
reconnaît que les descriptions pathologi-
ques et cliniques de la dégénérescence
cortico-basale ne correspondent pas
entièrement. Il serait donc utile de faire
une distinction entre le syndrome apra-
xique extrapyramidal qui se manifeste
par des signes cliniques, le syndrome de
la dégénérescence cortico-basale et l’en-
tité pathologique de la dégénérescence
cortico-basale (voir ci-dessous).
Démence reliée à une SLA
Depuis peu, les chercheurs portent un
grand intérêt à la relation entre la
démence et la SLA. Dans un premier
temps, cette relation a été décrite dans la
maladie de Creutzfeldt-Jakob, mais il
semble aujourd’hui que plusieurs de ces
patients n’étaient pas atteints d’une ma-
ladie à prion, mais plutôt d’une
dégénérescence fronto-temporale s’ac-
compagnant de changements spongi-
formes des couches superficielles du
cortex. D’ailleurs, un nombre croissant
d’articles sont publiés sur cette question,
touchant, par exemple, des patients
atteints d’une dégénérescence fronto-
temporale et d’une aphasie progressive
primaire et présentant une SLA ainsi que
des cas de SLA s’accompagnant d’une
démence. Des auteurs ont récemment
laissé entendre que des cas de dégé-
nérescence fronto-temporale accom-
pagnée d’une SLA présentent des inclu-
sions neuronales spécifiques, négatives
pour les protéines tau et synucléine et
positives pour l’ubiquitine. Néanmoins,
la spécificité de ces caractéristiques a été
remise en question par plusieurs descrip-
tions de cette pathologie ne s’accompa-
gnant pas d’une SLA et de plusieurs
autres cas de maladie de Pick ou de
dégénérescence fronto-temporale ac-
compagnée d’une SLA, mais en l’ab-
sence d’inclusions ubiquitine-positives.
Formes neuropathologiques
Il y a peu de temps encore, la présence
ou l’absence de corps de Pick et de neu-
rones gonflés ainsi que leur distribution
servait à la classification de sous-
groupes. La coloration différentielle à
l’aide d’épitopes phosphorylés (tau,
ubiquitine, cristalline αB et Gallyas) a
permis de distinguer, plus ou moins
clairement, les formes de pathologie
suivantes :
1) Démence accompagnée de corps de
Pick, définie par la présence de corps
de Pick argyrophiles, tau-immuno-
réactifs, dans le gyrus dentatus de
l’hippocampe ainsi que dans d’autres
territoires néocorticaux et sous-corti-
caux présentant des neurones gonflés
(cellules de Pick), une gliose et des
altérations spongiformes dans les
couches II et III du cortex (maladie
de Pick);
2) Gliose et perte de neurones, avec ou
sans spongiose, ou présence de neu-
rones gonflés dans les couches pro-
fondes, forme également appelée
« démence sans histologie distinc-
tive »;
3) Dégénérescence cortico-basale ca-
ractérisée par des neurones gonflés,
des plaques astrocytaires Gallyas-po-
sitives et tau-immunoréactives, des
filaments argyrophiles dans la sub-
stance blanche, le cortex et les
noyaux gris, et une globose ou amas
neurofibrillaires en anneaux dans le
locus niger (inclusions cortico-basa-
les) (dégénérescence cortico-basale);
4) Inclusions cytoplasmiques, tau et
synucléine-négatives, ubiquitine-
La revue canadienne de la maladie d’Alzheimer • Mars 2001 • 7
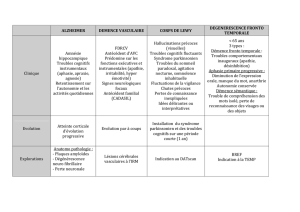
Figure 1 Le concept unificateur du complexe de Pick
Syndrome clinique du complexe de Pick
Démence
frontale
Aphasie progressive
primaire
Dégénérescence
cortico-basale
SLA
SLA
SLA : sclérose latérale amyotrophique (peut se manifester
dans l’une ou l’autre des composantes de ce complexe).

positives dans le gyrus dentatus et
d’autres régions corticales et sous-
corticales, avec ou sans SLA, telles
qu’elles sont décrites ci-dessus.
Puisque ces différentes formes se
chevauchent du point de vue des ca-
ractéristiques morphologiques et de leur
distribution et qu’elles ne sont pas spéci-
fiques d’aucun phénotype clinique, il est
encore trop tôt pour les considérer
comme des entités distinctes.
Complexe de Pick
On a suggéré le terme « complexe de
Pick » pour éliminer la confusion qui
persiste lorsqu’on parle de maladie de
Pick et de démence fronto-temporale.5
Le complexe de Pick est un concept qui
permet d’unifier les syndromes cliniques
de démence fronto-temporale, d’aphasie
progressive primaire et de syndromes de
dégénérescence corticobasale qui se
chevauchent ainsi que leurs caractéris-
tiques neuropathologiques sous-jacentes;
ce concept met l’accent sur les points
communs plutôt que sur les différences.
Il tient compte à la fois du chevauche-
ment des caractéristiques pathologiques
et cliniques, il évite de restreindre la
pathologie et la symptomatologie cli-
nique au cortex fronto-temporal et il
reconnaît la relation avec la maladie de
Pick (Figure 1). Les termes « dégénéres-
cence fronto-temporale » et « démence
fronto-temporale » ne tiennent pas
compte d’une atteinte sous-corticale
souvent présente, de la pathologie parié-
tale et de la symptomatologie extrapyra-
midale ni de la relation avec la SLA. Qui
plus est, le terme « démence fronto-tem-
porale » sème la confusion parce qu’il
désigne l’aspect trouble du comporte-
ment de ce syndrome ou le syndrome
dans son ensemble, y compris l’aphasie
progressive primaire. Une confusion
semblable était causée par l’utilisation du
terme « maladie de Pick », expliquant
pourquoi ces deux affections n’étaient
pas diagnostiquées dans tous les cas.
Traitement
Le traitement de la maladie de Pick n’est
pas encore défini. On avait d’abord cru
que le métabolisme du zinc était anormal
chez ces patients, mais le traitement à
l’aide d’un chélateur s’est révélé ineffi-
cace. Plus récemment, l’expérience a
montré que le traitement symptomatique
de la nervosité, des comportements com-
pulsifs à l’aide d’inhibiteurs sélectifs du
recaptage de la sérotonine (ISRS) ou de la
trazodone chez des sujets atteints d’une
démence fronto-temporale a été utile. Le
traitement par le lithium a été essayé chez
quelques patients parce qu’on croyait que
cet agent influait sur la déphosphorylation
de la protéine tau, mais les résultats ont
été décevants.
Génétique
Des résultats de recherches récentes
mettant en évidence une relation géné-
tique avec le chromosome 17 q21-22
dans plusieurs familles nombreuses
atteintes d’un trouble ressemblant
beaucoup au complexe de Pick ont été
décrits.9La région commune du chromo-
some à tous ces sujets contient le gène
pour la protéine tau qui stabilise le micro-
tubule. Des chercheurs ont identifié plus
de 20 mutations de la protéine tau et
diverses caractéristiques phénotypiques.
Cette découverte génétique apporte un
argument puissant en faveur de l’unité de
ce syndrome et évoque une pathogenèse
possible. Des études biochimiques
récentes sur le fractionnement de la pro-
téine tau permettraient d’expliquer
quelques-unes des variations de la
pathologie et des manifestations cli-
niques, mais il est encore trop tôt pour
relier les sous-types à certains tableaux
cliniques. La cartographie génique, des
études biochimiques et histochimiques
aideront à mieux comprendre ce syn-
drome, mais on ne doit pas perdre de vue
la cohérence clinique, pathologique et
génétique et on doit se montrer prudent
dans l’interprétation des différences.
Résumé
Le diagnostic de démence fronto-tempo-
rale, ou maladie de Pick, est évoqué chez
un patient, en général âgé de moins de
70 ans, qui présente des antécédents de
désinhibition, de démence « frontale » ou
d’aphasie progressive primaire et chez qui
la neuroimagerie révèle une atrophie fron-
to-temporale. Ces patients devraient être
orientés vers un centre de soins tertiaires.
8• La revue canadienne de la maladie d’Alzheimer • Mars 2001
Références
1. Pick, A. Über die Beziehungen der senilen
Hirnatrophie zur Aphasie. Prag Med
Wchnschr, 1892; 17:165-7.
2. Constantinidis, J, Richard, J, Tissot, R. Pick’s
disease - histological and clinical correla-
tions. Europ Neurol, 1974; 11:208-17.
3. The Lund and Manchester Groups. Clinical
and neuropathological criteria for fron-
totemporal dementia. J Neurol Neurosurg
Psychiatry, 1994; 57:416-8.
4. Mesulam, MM. Slowly progressive aphasia
without generalized dementia. Ann Neurol,
1982; 11:592-8.
5. Kertesz, A, Hudson, L, Mackenzie, IRA, et
coll. The pathology and nosology of prima-
ry progressive aphasia. Neurology, 1994;
44:2065-72.
6. Snowden, JS, Goulding, PJ, Neary, D. Seman-
tic dementia: a form of circumscribed cere-
bral atrophy. Behav Neurol, 1989; 2:167-82.
7. Rebeiz, JJ, Kolodny, EH, Richardson, EP Jr.
Corticodentatonigral degeneration with
neuronal achromasia. Arch Neurol, 1968;
18:20-33.
8. Gibb, WRG, Luthert, PJ, Marsden, CD.
Corticobasal degeneration. Brain, 1989;
112:1171-92.
9. Wilhelmsen, K. Frontotemporal dementia is
on the MAPt. Ann Neurol, 1997; 41:139-
40.
Le complexe de Pick est un concept qui permet d’unifier les
syndromes cliniques de démence fronto-temporale, d’apha-
sie progressive primaire et de syndromes de dégénérescence
cortico-basale qui se chevauchent ainsi que leurs caractéris-
tiques neuropathologiques sous-jacentes; ce concept met
l’accent sur les points communs plutôt que sur les
différences.
1
/
4
100%