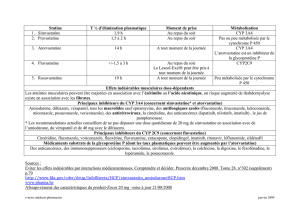
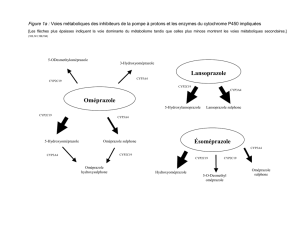
Lire l`article complet

12
La Lettre de l’Infectiologue - Tome XV - n° 1 - janvier 2000
MiSE AU POINT THÉRAPEUTIQUE
élimination est une étape clé de la pharmacocinétique
des médicaments. La vitesse d’élimination a un
impact direct sur la clairance et donc la demi-vie et
les modalités d’administration d’un médicament. En bref, deux
grandes voies participent à l’élimination des médicaments :
– l’élimination sous forme inchangée, par excrétion rénale
(et/ou biliaire) ;
– les réactions de biotransformation ou métabolisme, qui impli-
quent des enzymes de phase I (oxydation) ou de phase II (conju-
gaison) et conduisent à la formation de métabolites excrétés par
le rein ou la bile.
Les médicaments peuvent donc être éliminés en totalité par voie
rénale sous forme inchangée (par exemple les aminosides, ou
la majorité des pénicillines ou céphalosporines), sous forme de
métabolites pour les médicaments lipophiles (macrolides...), ou
de façon mixte. Il est de plus en plus évident que la connais-
sance des enzymes impliquées dans le métabolisme éventuel
est un facteur de réussite thérapeutique, puisqu’elle fournira
des informations sur la variabilité des concentrations, sur
l’effet de premier passage, sur les modifications pharmaco-
cinétiques éventuelles chez les patients insuffisants hépatiques,
et permettra de prévoir certaines interactions médicamenteuses.
Depuis les années 60, une hémoprotéine a été identifiée comme
la partie “active” des enzymes catalysant les réactions d’oxy-
dation, les mono-oxygénases. Cette hémoprotéine, qui fixe le
monoxyde de carbone et absorbe à 450 nm, a été appelée cyto-
chrome P450 (1). Depuis, plusieurs isoformes ont été identi-
fiées, chacune ayant des caractéristiques et des spécificités de
substrat différentes. Leur rôle dans le métabolisme des médi-
caments, en particulier des anti-infectieux, et les facteurs pou-
vant moduler leur activité vont être précisés.
DÉFINITION ET PRÉSENTATION DU (DES) CYTOCHROME(S) P450
Structure et rôle
Les cytochromes P450 (CYP) ont pour fonction l’oxydation
des molécules au moyen de réactions chimiques très diverses.
Ces réactions donnent naissance à des métabolites inactifs,
actifs ou toxiques. Les CYP sont des hémoprotéines dont la
structure est schématisée figure 1. Les modalités de fonction-
nement des cytochromes P450 ont fait l’objet de nombreuses
revues (2, 3). En résumé, la partie héminique du CYP (de struc-
ture tétrapyrolique), avec un atome de fer à l’état ferreux (Fe2+),
est capable de fixer l’oxygène moléculaire et de l’activer. Cet
Le système des cytochromes P450 :
définition, rôle et implication
dans la pharmacocinétique des anti-infectieux
!
A.M. Taburet*, V. Furlan*
* Pharmacie clinique, CHU de Bicêtre, AP/HP, 94270 Le Kremlin-Bicêtre.
RÉSUMÉ.
Les cytochromes P450 (CYP) sont des hémoprotéines qui participent au métabolisme oxydatif de nombreux médicaments. En fonc-
tion de la structure de la protéine fixée à l’hème, différentes familles et sous-familles ont été identifiées chez l’homme. Les principales formes
sont CYP 1A2, 2D6, 2C9, 2C19, 2E1 et 3A4. Certains sujets présentent un déficit en CYP 2D6, 2C9 et 2C19 (“métaboliseurs lents”). C’est le
CYP 3A4 qui, chez l’homme, est quantitativement le plus important : il représente en moyenne 30 % (avec une variabilité interindividuelle
importante) du contenu hépatique en cytochromes, et est également présent au niveau intestinal. Il est impliqué dans le métabolisme de plus
de 50 % des médicaments commercialisés. L’activité des CYP peut être modifiée sous l’action de substances inductrices telles que la rifampi-
cine, le phénobarbital, la phénytoïne et la carbamazépine et des hydrocarbures polycycliques (qui augmentent la synthèse et donc la quantité
de CYP disponible), ou inhibitrices, qui vont provoquer une diminution du métabolisme et donc une augmentation des concentrations du ou
des deux médicament(s) associé(s). L’effet inducteur est maximal après 6 à 10 jours de traitement. Les inhibiteurs sont nombreux :les inhibi-
teurs puissants du CYP 3A4 sont l’érythromycine, le kétoconazole, l’itraconazole et le ritonavir. L’effet inhibiteur est immédiat. Des substances
non médicamenteuses sont susceptibles d’inhiber certains CYP. Par exemple, le jus de pamplemousse inhibe le CYP 3A4 localisé au niveau de
l’épithélium intestinal.
Mots-clés :
Cytochrome P450 - Interactions médicamenteuses - Induction - Inhibition.
L
’

oxygène activé est ensuite transféré sur le médicament fixé sur
la partie protéique du CYP, qui forme une “poche”, en regard
de l’atome de fer alors à l’état ferrique (Fe3+). Cette réaction
nécessite du NADPH et des réductases, qui vont transformer
l’atome de fer de l’état ferrique à l’état ferreux pour que le cycle
d’oxydation puisse à nouveau fonctionner (2, 3). La structure
de la chaîne protéique est variable et permet de distinguer plu-
sieurs isoenzymes du CYP, dont certaines sont caractérisées et
clonées. Elles sont classées en familles et sous-familles, en fonc-
tion de l’homologie de structure (4).
Chez l’homme adulte, les trois principales familles de cyto-
chromes impliquées dans le métabolisme de nombreux médi-
caments sont les CYP 1, 2 et 3. Les sous-familles sont les
CYP 1A, CYP 2A, CYP 2B, CYP 2C, CYP 2D, CYP 2E et
CYP 3A. Soixante-dix pour cent des cytochromes hépatiques
humains sont constitués des sept isoenzymes suivantes :
CYP 1A2, 2A6, 2B6, 2C, 2D6, 2E1 et 3A (2).
Substrats
Les principaux médicaments, substrats des différentes isoen-
zymes du CYP, sont énumérés dans le tableau I. Seuls sont
indiqués les médicaments pour lesquels une isoforme du CYP
participe à la majorité du métabolisme. Compte tenu des che-
vauchements de spécificité, certains médicaments sont méta-
bolisés par plusieurs CYP. Ainsi, par exemple, le propranolol
est métabolisé par les 2D6, 1A2 et 2C19, ou le diazépam par
les CYP 2C19 et 3A. Pour la warfarine, il existe une énantio-
sélectivité puisque les isomères R et S ne sont pas métabolisés
par les mêmes isoformes : l’isomère R est métabolisé par
les CYP 1A2, 3A et 2C19, l’isomère S est métabolisé par le
2C9 (5).
On constate que les médicaments métabolisés par le CYP 3A
sont nombreux ; il a été estimé que plus de la moitié des médi-
caments utilisés en thérapeutique humaine sont métabolisés par
cette famille de cytochromes P450.
Dans le domaine des médicaments anti-infectieux, les CYP par-
ticipent à l’élimination de certaines classes pharmacologiques :
les macrolides, les antifongiques azolés, les inhibiteurs de la
protéase du VIH et certains antiparasitaires. En revanche, les
aminosides, les pénicillines, les céphalosporines et de nom-
breux antiviraux sont excrétés en quasi-totalité par voie rénale
sous forme inchangée. Les cytochromes impliqués dans l’éli-
mination des principales classes d’anti-infectieux sont résumés
tableau II.
La Lettre de l’Infectiologue - Tome XV - n° 1 - janvier 2000
13
MiSE AU POINT
THÉRAPEUTIQUE
Figure 1. Représentation schématique du cytochrome P450 [CYP]
(2). L’hème est fixé à la partie protéique, au niveau de l’atome de
soufre d’une molécule de cystéine. C’est l’atome de fer qui, en pas-
sant de l’état ferreux (FE
2+
) à l’état ferrique (FE
3+
), participe au
cycle oxydatif. La partie protéique confère à chaque famille et sous-
famille une spécificité de fixation du substrat.
Tableau I. Liste (non exhaustive) des principaux CYP identifiés
chez l’homme et leurs substrats médicamenteux (5).
CYP SUBSTRAT
1A2 Théophylline*, tacrine, fluvoxamine*, clozapine*
2C9 Tolbutamide, AINS (diclofénac, ibuprofène, flurbiprofène,
indométacine, piroxicam*), phénytoïne, warfarine*, ticlopidine
2C19 Méphénytoïne, oméprazole*, lansoprazole, diazépam*, proguanil
2D6 Débrisoquine, dextrométorphan, fluoxétine*, maprotiline,
paroxétine, trazodone, venlafaxine, chlorpromazine, rispéridone,
thioridazine, tramadol*, flécaïnide, propafénone, métoprolol,
timolol*, nortriptyline, fluvoxamine*, perphénazine*
2E1 Halothane, théophylline*, éthanol*
3A !Analgésiques et apparentés : alfentanil, fentanil, méthadone*
!Antiépileptique : carbamazépine*
!Anti-infectieux : clarithromycine, érythromycine, itraconazole,
kétoconazole, miconazole, quinine, indinavir, nelfinavir, ritona-
vir, saquinavir, névirapine, éfavirenz, delavirdine, rifabutine*
!Anticancéreux : cyclophosphamide*, étoposide, ifosfamide*,
tamoxifène
!Cardiovasculaire :
"Antiarythmiques : amiodarone, disopyramide, xylocaïne,
quinidine
"Inhibiteurs calciques : nifédipine, nicardipine, diltiazem,
vérapamil
"Hypolipémiants : (lovastatine**), simvastatine, atorvastatine,
cérivastatine*
!Antihistaminiques : astémizole, loratadine
!Sphère gastro-intestinale : cisapride
!Immunosuppresseurs : ciclosporine, tacrolimus,
sirolimus (rapamycine)
!Antidépresseurs : sertraline
!Neuroleptiques : pimozide
!Sédatifs : alprazolam, midazolam, triazolam, zolpidem*
!Hormones : dexaméthasone, éthinylestradiol, fénastéride,
flutamide, méthylprednisolone, prednisone, testostérone
!Autres : alcaloïdes de l’ergot de seigle, sildénafil
* Lorsque des médicaments sont métabolisés par plusieurs CYP, seul(s) le(s)
principal(aux) est (sont) mentionné(s).
** Non commercialisée en France.
FeV
S
O
protéine
Site de fixation
du substrat

Localisation
C’est au niveau hépatique que la majorité des CYP sont iden-
tifiés. Ils sont situés dans le réticulum endoplastique, retrouvé
dans la fraction microsomale lors du fractionnement cellulaire.
Chez l’homme, la distribution tissulaire des CYP est ubiqui-
taire. Ils sont prédominants dans le foie, mais aussi présents
dans d’autres organes tels que les reins, les poumons, le cer-
veau, la peau, l’intestin et le placenta (2). Les isoformes du
CYP 3 (CYP 3A4 et 3A5) et du CYP 2C (2C8, 2C9, 2C18 et
2C19) sont les plus abondantes. L’expression de ces protéines
est variable en fonction des tissus et des espèces, et il existe une
variabilité interindividuelle importante. Elles représentent res-
pectivement en moyenne 30 % et 20 % du contenu hépatique
en cytochromes ; le CYP 1A2 représente 13 % et le CYP 2D6
10 % (2).
La présence de CYP 3A au niveau intestinal contribue à aug-
menter l’élimination présystémique et à diminuer la biodispo-
nibilité de certains médicaments. De plus, comme le montre
la figure 2, un transporteur, la glycoprotéine P (P-GP), est asso-
cié au CYP 3A des entérocytes et participe à l’élimination
présystémique en faisant sortir le médicament de la cellule qui,
réabsorbé, peut à nouveau être métabolisé par le CYP 3A (6).
Cette “recirculation” augmente le métabolisme intestinal puis-
qu’elle évite la saturation du CYP 3A. Les substrats du CYP 3A
et de la P-GP sont souvent identiques (7).
FACTEURS PHYSIOPATHOLOGIQUES MODIFIANT L’ACTIVITÉ
De nombreux facteurs peuvent moduler l’activité des CYP chez
l’homme, entraînant ainsi des variations du métabolisme des
médicaments, et donc de leur efficacité ou de leur toxicité. Les
conséquences cliniques vont dépendre de trois éléments prin-
cipaux : l’importance de la voie des CYP dans l’élimination
globale du médicament, la concentration active du produit
(molécule mère et/ou métabolite) au site d’action et la marge
thérapeutique. Plus cette dernière sera faible, plus il faudra être
vigilant.
Génétique (2)
Le polymorphisme génétique contribue largement aux varia-
tions interindividuelles de la réponse à certains médicaments.
Il concerne trois principales isoenzymes : CYP 2A6, 2D6 et
2C19. La diminution importante, voire l’absence, d’activité que
l’on peut noter chez les “métaboliseurs lents” entraîne une aug-
mentation des concentrations sanguines et de la réponse phar-
maco-toxicologique. Ainsi, 5 à 8 % de la population occiden-
tale d’origine caucasienne n’exprime pas le CYP 2D6, alors
que la prévalence est de 1 % chez les Orientaux. À l’inverse,
le phénotype “métaboliseurs lents” du CYP 2C19 est plus fré-
quent chez les Orientaux (# 20 %) que chez les Caucasiens
(< 5 %). Les conséquences cliniques dépendent de la marge
thérapeutique du médicament, de l’activité du métabolite formé
et de la participation d’autres isoformes du CYP au métabo-
lisme. Le déficit en CYP 2C9 semble plus rare (< 1 %).
Âge
Il existe des variations qualitatives et quantitatives des CYP au
cours du développement. Cela est particulièrement net chez le
fœtus humain, pour lequel certaines isoenzymes sont absentes
14
La Lettre de l’Infectiologue - Tome XV - n° 1 - janvier 2000
MiSE AU POINT THÉRAPEUTIQUE
Tableau II. Rôle des CYP dans l’élimination des anti-infectieux
(5).
Famille Médicament Fraction CYP
antimicrobienne métabolisée impliqué
(%)
Macrolides Érythromycine 90 3A
Clarithromycine > 70 3A
Azithromycine 30-35 3A ?
Josamycine 40-50 ? 3A
Roxithromycine 50 3A
Fluoroquinolones Ciprofloxacine 20 ?
Imidazolés Itraconazole > 90 3A
Kétoconazole > 90 3A
Fluconazole 11 3A
Rifamycines Rifabutine 90 3A > déacétylase
Rifampicine 60-65 3A
Antiparasitaires Dapsone 70-85 NAT* > 3A
Proguanil > 50 2C9 > 3A
Quinine > 80 3A (> 50 %)
Halofantrine 3A
Antiprotéases Ritonavir > 90 3A > 2D6
Saquinavir > 90 3A
Indinavir 80 3A (majeur)
Nelfinavir 90 3A > 2C19
Amprénavir 90 3A
* NAT : N-acétyltransférase.
Figure 2. Schéma du rôle du CYP 3A et de la glycoprotéine P (P-
GP) dans l’effet de premier passage.
P-GP : glycoprotéine P
D : médicament
m : métabolite
D
D
D
D
D
D
D
D
mVeine porte
Entérocytes
Lumière intestinale
CYP 3A4
m
FOIE
D
CYP 3A4
P-GP
D
P-GP CYP 3A4

(CYP 2C), alors que d’autres sont présentes, mais absentes chez
l’adulte (CYP 3A7). C’est pourquoi certains médicaments éli-
minés plus lentement chez le nouveau-né ont une demi-vie
allongée dans la période néonatale (diazépam, midazolam, théo-
phylline). Ce phénomène est encore plus net chez les nouveau-
nés prématurés (8, 9).
Pathologies
Les pathologies peuvent aussi, à des degrés divers, modifier le
métabolisme des médicaments. C’est le cas des pathologies thy-
roïdiennes, du diabète, de l’insuffisance rénale et surtout de
l’insuffisance hépatique (particulièrement les cirrhoses sévères
et les hépatites). Dans cette dernière, il a été rapporté des dimi-
nutions des quantités des CYP de 50 %, 80 % et 100 % res-
pectivement pour les CYP 2C, CYP 3A4 et le CYP 2E1 (10) ;
ces chiffres sont très variables en fonction de la sévérité de
l’atteinte hépatique.
SUBSTANCES MODIFIANT L’ACTIVITÉ DES CYP :
INTERACTIONS
L’action d’une autre substance médicamenteuse ou d’une sub-
stance non médicamenteuse (substance de l’environnement, ali-
mentaire...) sur le métabolisme d’un médicament est classi-
quement divisée en réactions d’induction ou d’inhibition. Leurs
caractéristiques et leurs conséquences sont résumées dans
le tableau III. Une substance inductrice augmente la vitesse
du métabolisme du médicament associé, alors qu’une substance
inhibitrice la diminue. Si l’effet d’une inhibition est très sou-
vent immédiat, surtout s’il s’agit de compétition entre deux sub-
strats, l’effet de l’induction qui nécessite une synthèse protéique
n’est maximal qu’après quelques jours de coadministration des
deux substances (en général 6 à 10 jours). À l’inverse, à l’arrêt
du traitement, l’inhibition est rapidement réversible sauf si la
substance inhibitrice a une demi-vie longue, alors que l’effet
de l’induction disparaîtra en fonction du turn-over du CYP
induit et de la demi-vie de la substance inductrice (11, 12).
Facteurs environnementaux
Les substances de l’environnement qui agissent en inhibant ou
induisant les CYP contribuent à en augmenter la variabilité.
!Inducteurs (12).Certaines substances alimentaires peuvent
agir comme des inducteurs enzymatiques, tels les indols (conte-
nus dans le chou), qui augmentent l’activité du 1A2, et
l’éthanol (en absorption chronique), qui induit le CYP 2E1. À
noter que l’éthanol en ingestion aiguë est inhibiteur du
CYP 2E1.
Les hydrocarbures polycycliques contenus dans la fumée de
cigarette ou les aliments cuits au charbon de bois sont des induc-
teurs connus du CYP 1A2. La consommation de cigarettes est
donc un facteur à prendre en compte lors d’un traitement par
des médicaments métabolisés par ce CYP, surtout si la marge
thérapeutique est faible, comme c’est le cas de la théophylline.
!Inhibiteurs. Il est probable que certaines substances endo-
gènes, ou de l’alimentation, ont des propriétés inhibitrices sur
les CYP, sans jamais avoir été identifiées. Un exemple décou-
vert fortuitement est la propriété inhibitrice du jus de pample-
mousse sur le CYP 3A (13). Il est probable que le (les) com-
posé(s) inhibiteur(s) administré(s) par voie orale ne parviennent
pas jusqu’au foie, ce qui fait que le jus de pamplemousse semble
être un inhibiteur assez spécifique du CYP 3A localisé dans les
entérocytes. L’effet inhibiteur est important pour les inhibiteurs
calciques (félodipine), plus modeste pour la ciclosporine.
L’effet est transitoire et, pour avoir un effet maximal, il est
nécessaire d’ingérer le médicament et le jus de pamplemousse
simultanément. C’est ainsi que la biodisponibilité du saquina-
vir, médicament fortement métabolisé par le CYP 3A intesti-
nal, double en présence de jus de pamplemousse (13).
Facteurs pharmaco-toxicologiques (12-14)
!Les médicaments inducteurs enzymatiques. Les médica-
ments inducteurs les plus puissants sont la rifampicine, le phé-
nobarbital, la phénytoïne et la carbamazépine. Ils induisent sur-
tout les CYP 3A et 2C. L’isoniazide induirait spécifiquement
le CYP 2E1. Le caractère inducteur puissant de la rifampicine
est un élément important à prendre en compte. Ainsi, la rifam-
picine est contre-indiquée avec les inhibiteurs de protéase, car
elle en diminue de façon très importante les concentrations, qui
se situent alors en dessous du seuil d’efficacité antivirale. D’une
façon générale, il y a lieu d’être vigilant lorsqu’on associe la
rifampicine à des médicaments métabolisés en grande partie
par le CYP 3A. D’autres médicaments comme la rifabutine, la
névirapine ou l’éfavirenz sont des inducteurs plus modestes.
Leur administration peut nécessiter d’augmenter la posologie
du médicament associé. Récemment, les associations névira-
pine-indinavir ou rifabutine-indinavir en sont des exemples. La
névirapine diminue les concentrations d’indinavir d’au moins
30 % en moyenne par induction de son métabolisme. Il est
nécessaire chez certains patients d’augmenter la posologie de
l’indinavir à 1 g x 3 (vs 800 mg x 3) pour conserver un niveau
de concentration efficace (15).
La Lettre de l’Infectiologue - Tome XV - n° 1 - janvier 2000
15
MiSE AU POINT
THÉRAPEUTIQUE
Paramètre Modification observée avec
Inducteur Inhibiteur
!Mécanisme Augmentation Compétition (réversible)
synthèse enzyme ou fixation irréversible
!Délai d’action Maximal Immédiat
en 6-10 jours
!Conséquences
pharmacocinétiques
– Sur les concentrations Diminution Augmentation
plasmatiques
– Sur la production Augmentée Diminuée
de métabolites
– Sur la clairance Augmentation Diminution
!Conséquences Diminution Augmentation
thérapeutiques de l’effet thérapeutique de l’effet thérapeutique
(s’il existe une relation (sauf si métabolite actif) et/ou effets indésirables
concentration/effet)
Tableau III. Effets comparés des inducteurs et inhibiteurs enzy-
matiques (12).

Les médicaments antiépileptiques (phénobarbital, phénytoïne,
carbamazépine) doivent être manipulés avec précaution du fait
de leur propriété inductrice enzymatique.
!Les médicaments inhibiteurs enzymatiques
"Mécanisme de l’inhibition. Les médicaments responsables
d’inhibition enzymatique sont beaucoup plus nombreux. Les
inhibitions peuvent être compétitives ou non compétitives. L’in-
hibition compétitive ou réversible se caractérise par une simple
compétition entre le substrat et l’inhibiteur pour la même isoen-
zyme. La molécule qui présentera l’affinité la plus grande pour
le CYP inhibera le métabolisme de l’autre. De nombreuses
interactions de ce type sont décrites dans la littérature ; elles
concernent l’ensemble des CYP. Le métabolisme du tacrolimus
est inhibé par le kétoconazole (16). Il existe une interaction dia-
zépam-oméprazole impliquant le CYP 2C19 qui ne survient
que chez les métaboliseurs rapides, non déficients en
CYP 2C19. La quinidine est un inhibiteur puissant du CYP 2D6
et une interaction tolbutamide-phénytoïne (CYP 2C9) a été
décrite. L’inactivation irréversible est moins fréquente. Un
exemple bien étudié est celui de l’inhibition du CYP 3A par
certains macrolides (17) tels que l’érythromycine ou la clari-
thromycine. Un métabolite “nitroso” de l’érythromycine forme
un complexe stable avec le CYP 3A, empêchant les oxydations
ultérieures, tant que de nouvelles hémoprotéines du CYP 3A
ne sont pas synthétisées. Des macrolides comme la josamycine
et la roxithromycine ont une affinité moindre pour le CYP 3A,
et peu d’interactions sont décrites. Quant à l’azithromycine ou
la dirithromycine, elles n’ont pas à ce jour été impliquées dans
des interactions cliniquemement significatives.
Un certain nombre de médicaments identifiés comme étant des
inhibiteurs puissants sont présentés dans le tableau IV. On
remarquera le rôle de certains anti-infectieux (macrolides, anti-
fongiques azolés, inhibiteurs de la protéase du VIH).
"Conséquences thérapeutiques d’une inhibition (11, 12).
Dans la majorité des cas, l’inhibition d’un CYP entraîne une
augmentation de la concentration des molécules mères qu’il
métabolise, et un risque augmenté de toxicité. C’est ainsi que
le kétoconazole ou l’itraconazole, inhibiteurs puissants, élèvent
les concentrations de ciclosporine ou de tacrolimus de façon
importante, augmentant le risque de néphrotoxicité. C’est éga-
lement la raison pour laquelle il existe une contre-indication à
l’association entre les inhibiteurs puissants et des médicaments
à toxicité cardiaque : cisapride, astémizole, pimozide. Les inter-
actions décrites avec la terfénadine ont été responsables de son
retrait du marché. Ce médicament est en lui-même inactif, mais
possède une toxicité cardiaque ; c’est le dérivé acide de la ter-
fénadine formé par l’intermédiaire du CYP 3A qui possède l’ac-
tivité antihistaminique. L’inhibition du CYP 3A augmente les
concentrations de terfénadine, qui sont normalement indétec-
tables, et donc la toxicité. Cela a été décrit avec de nombreux
inhibiteurs du 3A4, dont l’érythromycine (11). Certaines qui-
nolones (énoxacine et ciprofloxacine) inhibent le CYP 1A2, et
sont responsables d’interactions avec la théophylline.
Lorsque le médicament et le métabolite sont actifs, l’activité
thérapeutique reste inchangée. C’est l’exemple de l’inhibition
de la transformation de la clarithromycine en 4-hydroxyclari-
thromycine par l’indinavir : les concentrations de clarithromy-
cine augmentent, mais celles du métabolite actif (la 4-hydroxy-
clarithromycine) diminuent. En revanche, lorsque la substance
mère est une prodrogue, l’activité thérapeutique peut diminuer,
c’est le cas du proguanil (seul le cycloguanil étant actif). Les
interactions décrites avec la terfénadine ont été responsables de
son retrait du marché.
Dans certains cas, les inhibiteurs sont utilisés pour augmenter
les concentrations d’un médicament. Ainsi, le ritonavir est uti-
lisé pour diminuer l’effet de premier passage du saquinavir (18).
Les concentrations augmentées de saquinavir sont alors très
supérieures à la concentration virale inhibitrice. Il a été égale-
ment démontré que cette inhibition diminue la variabilité inter-
individuelle des concentrations de saquinavir.
"Enfin, on a évoqué l’utilisation de certains médicaments
bien tolérés pour diminuer la posologie de certains médica-
ments onéreux, comme les immunosuppresseurs (19). Une telle
attitude n’est pas, à l’heure actuelle, recommandée en France.
!Prévision des interactions médicamenteuses
Il existe à l’heure actuelle des techniques in vitro bien validées,
qui permettent d’identifier le(s) CYP impliqué(s) dans le méta-
bolisme des nouvelles molécules, et de mettre en évidence les
inhibiteurs de ces CYP. Ces techniques font appel à des micro-
somes hépatiques humains ou des cellules transfectées. Les cel-
lules d’hépatocytes, qui permettraient en outre d’identifier des
propriétés inductrices, sont plus complexes à utiliser (5, 20).
Ces techniques se substituent aux modèles animaux, souvent
peu prédictifs dans le domaine du métabolisme. Elles sont
d’ailleurs citées dans la dernière directive de l’Agence euro-
péenne sur l’étude des interactions médicamenteuses (21).
CONCLUSION
Les progrès de la biologie moléculaire ont permis d’identifier
et de caractériser les CYP qui jouent un rôle fondamental dans
le métabolisme de nombreux médicaments.
16
La Lettre de l’Infectiologue - Tome XV - n° 1 - janvier 2000
MiSE AU POINT THÉRAPEUTIQUE
CYP Médicament
1A Fluvoxamine, énoxacine, ciprofloxacine
2D6 Quinidine, fluoxétine
3A Macrolides !TAO (non commercialisé),
érythromycine, clarithromycine
Antifongiques azolés !Kétoconazole, itraconazole
(à un moindre dégré fluconazole)
Antirétroviraux !Par ordre décroissant de puissance
inhibitrice : ritonavir +++ > indinavir,
nelfinavir, amprénavir ++
>saquinavir
Tableau IV. Les principaux inhibiteurs des CYP (5, 11, 12, 14).
 6
6
1
/
6
100%