article original Épidémiologie moléculaire de la mucoviscidose en

article original
Épidémiologie moléculaire de la mucoviscidose
en Tunisie
T. Messaoud
1
S. Bel Haj Fredj
1
A. Bibi
1
J. Elion
2
C. Férec
3
S. Fattoum
1
1
Laboratoire de biochimie clinique,
Hôpital d’Enfants de Tunis, Tunisie
2
Laboratoire de biochimie génétique,
Fédération de génétique,
hôpital Robert Debré, Paris, France
3
Laboratoire de génétique moléculaire,
Établissement français du sang –
Bretagne, Brest, France
Article reçu le 4 mars 2005,
accepté le 9 septembre 2005
Résumé.La mucoviscidose ou fibrose kystique du pancréas est la maladie
autosomique récessive la plus fréquente dans la population nord européenne.
Le développement des techniques de biologie moléculaire a largement facilité
la mise en place des techniques d’étude des différentes lésions moléculaires du
gène CFTR responsables de la mucoviscidose dont le nombre actuellement est
supérieur à 1 300 mutations. Dans le présent travail, nous rapportons la straté-
gie d’étude de ces lésions sur 390 enfants appartenant à 383 familles non
apparentées suspects de mucoviscidose et qui nous sont parvenues de la plupart
des régions de la Tunisie. Plusieurs techniques de biologie moléculaire ont été
utilisées pour le diagnostic génotypique : l’extraction de l’ADN à partir du
sang périphérique, la réaction de polymérisation en chaîne (PCR), l’électro-
phorèse sur gel de polyacrylamide, la PCR multiplex et l’électrophorèse sur gel
en gradient dénaturant (DGGE) suivie au besoin par une réaction de séquen-
çage du gène CFTR. Cette étude a permis d’identifier 17 mutations localisées
sur différents exons du gène CFTR. La mutation délétionnelle F508del est la
plus fréquente (50,74 %) suivie de 3 autres mutations communément retrou-
vées dans les pays du bassin méditerranéen : G542X, W1282X et N1303K, à
côté de 4 autres mutations (T665S, 2766del8, F1166C, L1043R) retrouvées
exclusivement à ce jour, dans la population tunisienne. Les résultats obtenus
ont permis d’une part, d’établir le profil des mutations mucoviscidosiques et
leur distribution dans le pays et, d’autre part, de mettre en œuvre une stratégie
de prévention de ces maladies à travers le conseil génétique et le diagnostic
prénatal.
Mots clés :mucoviscidose, défaut moléculaire, population tunisienne
Abstract.Cystic fibrosis is the most frequent autosomal recessive genetic
disease in North European population. This pathology seems to not be rare in
Tunisia. On another hand, development of molecular biology technics has
largely contributed to implement the study of the different mutations in the
CFTR gene where over of 1.300 mutations were reported. Herein, we describe
the strategy used to detect molecular defects responsible of cystic fibrosis on
390 childrens (383 families) in Tunisian population. Several technics were
performed for genotype diagnosis: DNA extraction was from peripheral blood.
Polymerase chain reaction (PCR) and polyacylamide gel electrophoresis, and
reverse dot blot procedures were used to detect known point mutations. Dena-
turant gradient gel electrophoresis (DGGE) were used in a next step searching
for the unknown point mutations that are later indentified by automated
sequencing on ABIprism 310. This strategy allowed us to detect 17 different
mutations located on the different exons of the CFTR gene. The most frequent
was the F508del (50,74 %) followed by three other mutations (G542X,
W1282X and N1303K) known to be common in the Mediterranean area. For
mutations (T665S, 2766 del8, F1166C, L1043R) were exclusively found, up to
Tirés à part : S. Fattoum
abc
Ann Biol Clin 2005 ; 63 (6) : 627-30
Ann Biol Clin, vol. 63, n° 6, novembre-décembre 2005 627
Copyright © 2017 John Libbey Eurotext. Downloaded by a robot coming from 88.99.165.207 on 25/05/2017.

now, in the Tunisian population. Our results permitted to establish cystic fibro-
sis mutations and their distribution in Tunisia and to implement an appropriate
prevention program of these diseases through the genetic council and prenatal
diagnosis.
Key words:cystic fibrosis, molecular defect, Tunisian population
La mucoviscidose est la maladie héréditaire autosomique
récessive la plus fréquente dans les populations caucasien-
nes d’Europe centrale (1/3 000). Elle est réputée rare en
Afrique du Nord et au Maghreb. C’est une exocrinopathie
généralisée, frappant les glandes séreuses et les glandes à
sécrétion muqueuse. Sont touchés principalement l’appa-
reil respiratoire, le tube digestif, mais également les glan-
des sudoripares et le tractus génital avec obstruction des
canaux déférents entraînant une stérilité par azoospermie
[1]. Son diagnostic a toujours reposé sur des signes clini-
ques d’orientation (pulmonaires et ou digestifs) complété
par un test de la sueur positif avec des chlorures ≥60
mmol/L.
La confirmation du diagnostic se fait par l’analyse molé-
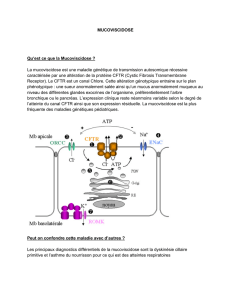
culaire du gène CFTR à la recherche de la lésion en cause.
Le gène CFTR est porté par le bras long du chromosome
7 ; il code pour une protéine de 1 480 acides aminés,
appelée CFTR (cystic fibrosis transmembrane conduc-
tance regulator) responsable du transfert des ions chloru-
res à travers la membrane cellulaire [2]. Plus de 1 300
mutations différentes responsables de mucoviscidose ont
été identifiées de par le monde. La mutation F508del reste
la plus fréquente en Europe [3]. Ces mutations sont
regroupées en plusieurs classes selon la nature de la lésion
moléculaire et le mécanisme physiopathologique en
cause. Cette classification est à la base de l’étude des
corrélations génotype-phénotype dans l’expression de la
mucoviscidose.
Nous rapportons dans le présent travail une mise à jour de
la fréquence et de la distribution des mutations identifiées
concernant une cohorte de 383 familles qui nous sont
parvenues de la plupart des régions de la Tunisie pour
suspicion de mucoviscidose.
Matériel et méthodes
Population
En Tunisie, les premiers travaux ont commencé en 1990
avec l’analyse du test de la sueur, que nous avons réalisé
chez 5 571 enfants suspects de mucoviscidose, l’âge de
ces malades varie entre 1 mois et 12 ans. Ce test était
positif dans 7 % des cas soit 390 enfants appartenant à 383
familles non apparentées. Le dosage des ions chlorures est
effectué par potentiométrie sur de la sueur recueillie par
iontophorèse [4, 5]. Les taux usuels sont généralement
admis comme : normal < 40 mmol/l et douteux entre 40 et
60 mmol/L. L’analyse moléculaire a été effectuée chez les
sujets présentant des signes cliniques évocateurs de muco-
viscidose avec au moins deux tests de la sueur positifs
(269 enfants). Devant la sévérité du tableau clinique, deux
enfants ayant présenté un test négatif ont bénéficié d’une
analyse génétique. De même, cette étude a été réalisée
chez un sujet d’origine algérienne présentant une stérilité
masculine.
Stratégie d’étude moléculaire
La stratégie d’étude moléculaire passe par plusieurs
étapes nécessitant une extraction de l’ADN [6], une
amplification par PCR simple [7] et une électrophorèse sur
gel de polyacrylamide pour la recherche des mutations
F508del et 2766del 8 [8]. Pour les mutations connues, une
PCR multiplex utilisant un mélange de plusieurs couples
d’amorces à la fois suivie d’une électrophorèse capillaire
sur analyseur automatique de fragments (ABI 310 Applied
BioSystem, États-Unis). Cette technique permet actuelle-
ment de tester 31 mutations à la fois (oligonucleotide lin-
gand assay : OLA) [9].
Pour les mutations inconnues, une électrophorèse sur gel à
gradient dénaturant (DGGE) est pratiquée pour compléter
l’exploration du gène CFTR [7, 10]. Une réaction de
séquençage selon la technique de Sanger permettra de pré-
ciser le siège et la nature des mutations ciblées par la
DGGE : E1104X, R74W, D1270N, Y122X, T665S,
R1066C, I148T, F1166C. Cette technique est réalisée sur
un automate de type ABI Prism 310 (Applied BioSystems,
États-Unis) [11]. Pour 40 malades, la chromatographie
liquide à haute pression dénaturante (dHPLC) a été réali-
sée en remplacement de la DGGE [12]. La recherche des
grandes délétions a été effectuée sur 32 malades par PCR
quantitative en temps réel.
Résultats et commentaires
Le test de la sueur effectué chez nos malades a été positif
chez 381 enfants avec des taux allant de 67 à 159 mmol/L
(moyenne des chlorures de 91,8 ± 24,4 mmol/L). Sur les
article original
Ann Biol Clin, vol. 63, n° 6, novembre-décembre 2005628
Copyright © 2017 John Libbey Eurotext. Downloaded by a robot coming from 88.99.165.207 on 25/05/2017.

540 chromosomes étudiés soit 270 malades, la lésion
moléculaire a été identifiée dans 70 % des cas révélant 17
mutations différentes (tableau 1).
Ces résultats confortent ceux rapportés précédemment sur
une étude préliminaire réalisée sur 47 chromosomes [13] ;
la mutation F508del est de loin la plus fréquente
(50,74 %) suivie de trois autres mutations communément
retrouvées dans les pays du pourtour méditerranéen :
G542X (7,96 %), W1282X (6,66 %) et N1303K (5,92 %).
La mutation 2766 del 8, nouvellement décrite en Tunisie
occupe la 5
e
place (4,25 %). Elle a été observée à l’état
homozygote dans deux cas et hétérozygote composite
dans 6 cas issus de six régions différentes du pays.
Les autres mutations identifiées sont plus rares : il s’agit
de la mutation 711+1G→T (4,23 %). La mutation
E1104X (1,85 %) nouvellement identifiée dans notre série
et 7 autres mutations encore plus rares : G85E, D1270N,
R74W, R1066C, Y122X, T665S et I148T.
La mutation V201M a été détectée par dHPLC [14], dans
un cas de stérilité masculine par agénésie bilatérale des
canaux déférents (CABVD). Par ailleurs, nous rapportons
deux nouvelles mutations à l’état hétérozygote, décrites
pour la première fois dans la littérature : la mutation
F1166C identifiée par DGGE et la mutation L1043R (par
dHPLC) et situées respectivement au niveau des exons 19
et 17a du gène CFTR.
L’homozygotie est retrouvée dans 64,44 % des cas dont
66,09 % sont F508del/F508del et 33,9 % non F508del/non
F508del. Ce phénomène est favorisé par le taux commu-
nément élevé de consanguinité en Tunisie, comme il a déjà
été observé dans d’autres maladies génétiques fréquentes
telles que la b-thalassémie et la drépanocytose. Dans notre
série la consanguinité a été notée à l’interrogatoire dans
211 couples, le degré de parenté concerne 86,44 % des cas
dont 28,81 % sont du 1
er
ou du 2
e
degré.
La répartition géographique a permis de constater que les
sujets étudiés sont originaires de 19 gouvernorats diffé-
rents (tableau 2). Le grand Tunis et le sud-est (SE) sem-
blent être les régions les plus concernées par la mucovisci-
dose avec respectivement 22,18 % et 18,18 % des patients
identifiés. Ceci peut être expliqué en partie par le recrute-
ment plus important de malades issus de ces régions. La
répartition des mutations permet de constater une grande
hétérogénéité dans la plupart des gouvernorats.
Enfin nous voulons noter la présence de deux cas de test
de la sueur négatifs (23 et 26 mmol/L) avec des signes
cliniques évocateurs qui nous ont poussé à procéder à
l’analyse moléculaire. Ceci nous a permis en effet d’iden-
tifier deux mutations à l’état homozygote dans les deux
cas (F508del/F508del et G542X / G542X).
Pour le restant des sujets présentant une forte suspicion
clinique de mucoviscidose (48 cas), aucune mutation n’a
été retrouvée par les techniques utilisées malgré un test de
la sueur positif à plusieurs reprises (de 92 à 113 mmol/L,
avec une moyenne de 97 ± 7,2 mmol/L).
La fréquence de 50,74 % pour la F508del peut cadrer avec
le gradient décroissant décrit pour cette mutation du nord-
ouest de l’Europe (88 % au Danemark) au sud-est [3]. Les
nouvelles mutations tunisiennes publiées en 1996 (T665S
et la 2766 del 8) et retrouvées dans 9 familles non appa-
rentées de diverses régions du pays n’ont pas encore été
rapportées dans d’autres ethnies. Cela suggère vraisem-
blablement une spécificité à la population tunisienne étu-
diée, d’autant plus que la mutation 2766 del 8 a été retrou-
vée dans la plupart des cas, en association avec la
mutation N1303K. Des études complémentaires sont
nécessaires pour un typage précis afin d’expliquer cette
hétérozgotie composite.
Tableau 1. Spectre des mutations du gène CFTR dans la
population tunisienne.
Mutations Nombre de chromosomes Fréquence
Homozygote Hétérozygote TotalPourcentage
F508del 230 44 274 50,74
G542X 34 9 43 7,96
W1282X 32 4 36 6,66
N1303K 20 12 32 5,92
711+ 1G→T 20 3 23 4,25
2766del 8 4 6 10 1,85
E1104X 4 5 9 1,66
G85E 4 2 6 1,11
R74W 2 2 0,37
D1270N 2 2 0,37
Y 122X 1 1 0,18
T665 S 1 1 0,18
R1066C 1 1 0,18
I 148T 1 1 0,18
F1166C 1 1 0,18
L1043R 1 1 0,18
V201M 1 1 0,18
Non déterminée 96 17,77
Tableau 2. Répartition géographique des familles répertoriées
pour mucoviscidose (1 ou 2 mutations identifiées).
Régions Familles
Nombre Pourcentage
Grand Tunis 60 22,18
Nord-Est 49 18,18
Nord –Ouest 25 9,30
Centre-Est 19 7,00
Centre-Ouest 26 9,50
Sud-Est 52 19,30
Étranger 1 0,37
Non déterminée 38 14,17
Épidémiologie moléculaire de la mucoviscidose
Ann Biol Clin, vol. 63, n° 6, novembre-décembre 2005 629
Copyright © 2017 John Libbey Eurotext. Downloaded by a robot coming from 88.99.165.207 on 25/05/2017.

Le taux élevé (17,77 %) de mutations restant non identi-
fiées après une première recherche ayant comporté l’ana-
lyse de tous les exons n’est pas pour nous étonner, cette
même proportion a été rapportée dans d’autres pays du
monde tels que l’Espagne, l’Italie et certaines régions de
la France. Le gène CFTR est en effet de grande taille
(250 Kb) et une recherche plus fine doit s’intéresser aux
zones de régulation du gène en 5’ et en 3’ mais également
aux séquences introniques non encore explorées. Nous
pensons en particulier à la mutation 1811+ 1,6 Kb A→G
retrouvée avec une fréquence élevée (8,8 %) dans les
régions du sud-ouest de la France avec lesquelles nous
partageons plusieurs autres mutations [15]. Nous pensons
également à une autre mutation, la 3120+1 G→A récem-
ment identifiée par DGGE à des taux significativement
importants (9 à 14 %) dans certaines populations arabes
(Arabie saoudite) ou d’origine africaine, avec lesquelles
d’ailleurs nous avons en commun une mutation très rare :
la D1270N généralement retrouvée dans les cas de stérilité
par agénésie des canaux déférents [16].
Conclusion
La mucoviscidose est une affection rare sans être excep-
tionnelle dans notre pays. Nous avons observé une nette
augmentation du nombre de patients diagnostiqués au fil
des années (prés de 383 familles répertoriées à ce jour)
grâce à la sensibilisation des praticiens et au developpe-
ment des moyens du diagnostic. Les données épidémiolo-
giques et moléculaires que nous venons d’évoquer sont de
nature à attirer l’attention sur la nécessité de ne pas
exclure cette pathologie de la routine des explorations
chez les enfants présentant des signes évocateurs de
mucoviscidose afin de permettre un diagnostic et une prise
en charge précoces. Elles ont également un intérêt majeur
dans l’étude des corrélations phénotype-génotype [17]
afin d’instaurer une prévention efficace de ces maladies
génétiques invalidantes, par le biais du diagnostic prénatal
chez les couples à risque.
Remerciements. Le présent travail a été mis en route dans
le cadre d’une coopération Tuniso-française (CNTS-Brest,
Hôpital Robert Debré-Paris) et est financé actuellement
dans le cadre d’un contrat recherche à l’hôpital d’enfants
de Tunis par les Ministères de la Recherche scientifique et
de la Santé publique (Lab santé 01). Nous remercions les
collègues du groupe d’hépatologie, gastroentérologie et
nutrition de la société tunisienne de pédiatrie qui ont bien
voulu nous adresser leurs patients pour étude génétique.
Références
1. Bienvenu T. Les bases moléculaires de l’hétérogénéité phénotypique
dans la mucoviscidose. Ann Biol Clin (Paris) 1997 ; 55 : 113-22.
2. Férec C, Mercier B, Audrézet MP. Les mutations de la mucovisci-
dose : du génotype au phénotype. Med Sci (Paris) 1994 ; 10 : 631-9.
3. Tomaiuolo R, Spina M, Castaldo G. Molecular diagnosis of cystic
fibrosis : comparison of four analytical procedures. Clin Chem Lab Med
2003 ; 41 : 26-32.
4. Gibson LE, Cooke RE. A test for concentration of electrolytes in
sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontopho-
resis. Pediatrics 1959 ; 23 : 545-9.
5. Marchand M, Jarreau C, Chauffert M, et al. Le test de la sueur. Ann
Biol Clin (Paris) 1998 ; 56 : 215-21.
6. Bienvenu T, Meunier C, Bousquet S, et al. Les techniques d’extraction
de l’ADN à partir d’un échantillon sanguin. Ann Biol Clin (Paris) 1999 ;
57 : 77-84.
7. Fisher SG, Lerman LS. DNA fragments differing by single base-pair
substitutions are separed in denaturing gels : correspondence with mel-
ting theory. Proc Natl Acad Sci USA 1983 ; 80 : 1579-83.
8. Messaoud T, Xerri B, Guemira F, Chniti F, Elion J, Fattoum S. Dia-
gnostic moléculaire de la mucoviscidose (DF508) en Tunisie (A propos
d’un cas familial). Revue maghrébine de pédiatrie 1992 ; 2 : 93-5.
9. Gomez-Liorente MA, Suarez A, Gomez-Liorente C, et al. Analysis of
31 CFTR mutation in 55 families from the south of spain. Early Hum
Dev 2001 ; 65 (Suppl.) : S161-S164.
10. Hayes VM, Wu Y, Osinga J, et al. Improvements in gel composition
analysis and electrophoretic conditions for broad range mutation analysis
by denaturing gradient gel electrophoresis. Nucleic Acids Res 1999 ; 27 :
29.
11. Sanger F, Nickler S, Coulson AR. DNA sequencing with chain termi-
nating inhibitors. Proc Natl Acad Sci USA 1977 ; 74 : 5463-7.
12. Xiao W, Oefner PJ. Denaturing high-performance liquid chromato-
graphy : a review. Hum Mutat 2001 ; 17 : 439-74.
13. Messaoud T, Verlingue C, Denamur E, et al. Distribution of CFTR
mutation in cystic fibrosis patients of tunisian origin : identification of
two novel mutations. Eur J Hum Genet 1996 ; 4 : 20-4.
14. Claustres M, Altieri JP, Guittard C, Templin C, Chevalier – Porst F,
Des Georges M. Are p.I 148 T, p.R.74 W and p.D 1270N cystic fibrisis
causing mutations? BMC Med Genet 2004 ; 5 : 19.
15. Frederici S, Iron A, Reboul MP, et al. CFTR gene analysis in 207
patients with cystic fibrosis in South west France : high frequency of
N1303K and 1811+ 1.6 Kb A→G mutations. Arch Pediatr 2001 ; 8 :
150-7.
16. Padoa C, Goldman A, Jenkins T, Ramsay M. Cystic fibrosis carrier
frequencies in populations of African origin. J Med Genet 1999 ; 36 :
41-4.
17. Dhouib K, Messaoud T, Dridi MF, et al. Contribution à l’étude des
corrélations génotype-phénotype dans la mucoviscidose. Revue tuni-
sienne de la santé militaire 2001 ; 3 : 326-30.
article original
Ann Biol Clin, vol. 63, n° 6, novembre-décembre 2005630
Copyright © 2017 John Libbey Eurotext. Downloaded by a robot coming from 88.99.165.207 on 25/05/2017.
1
/
4
100%