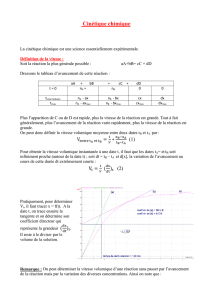
Spécificités de la cinétique chimique des systèmes

2005-06 Bernard Pieraggi 1
Cinétique Chimique
des
Systèmes Hétérogènes

2005-06 Bernard Pieraggi 2
La cinétique des réactions chimiques qui se produisent au sein des systèmes chimiques
hétérogènes présente des caractéristiques spécifiques. L'objet de ce cours est de
préciser ces spécificités, d'en développer les conséquences et de proposer une
méthodologie d'analyse des mécanismes et cinétiques des réaction hétérogènes.
Notions élémentaires de cinétique hétérogène
Introduction
Les applications industrielles de la cinétique hétérogène sont nombreuses et diverses.
Les exemples suivants sont seulement destinées à illustrer, de façon non exhaustive, la
variété des situations qui peuvent être rencontrées.
Le caractère hétérogène des systèmes considérés implique notamment que plusieurs des
processus et étapes réactionnelles se déroulent au niveau des interfaces qui délimitent et
séparent les diverses phases du système réactionnel. Le rôle et l'influence des étapes et
processus interfaciaux sont des éléments fondamentaux de la cinétique hétérogène.
Cette influence se traduit également par le rôle très important de la forme et de la taille
des réactifs et produits de réaction qui déterminent l'étendue des zones interfaciales et
leur éventuelle variation.

2005-06 Bernard Pieraggi 3
Notions élémentaires de cinétique hétérogène
Exemple 1 : Fabrication des circuits intégrés
Si monocristallin
SiO2amorphe
Si polycristallin
La fabrication des circuits intégrés nécessite de très nombreuses opérations : nettoyage,
dopage, gravage, oxydation, dépôts divers pour réaliser les architectures 3D des circuits
actuels…La micrographie ci-dessous illustre deux opérations importantes : la formation
des couches isolantes de SiO2et le dépôt de Si polycristallin.
Ces deux opérations, ainsi que le dopage des substrats
Si, résultent d'une combinaison, parfois très complexe,
de processus et étapes réactionnelles entre phases
gazeuse et solide.
Le contrôle de ces opérations exige une connaissance
approfondie et précise des mécanismes et cinétiques
des différentes étapes réactionnelles mises en œuvre.
Dépôt CVD de Si polycristallin
Oxydation du substrat Si
Dopage du substrat Si

2005-06 Bernard Pieraggi 4
Notions élémentaires de cinétique hétérogène
Exemple 2 : Désulfuration des gaz de combustion
Le soufre est présent dans de nombreux combustibles naturels, notamment le charbon et
divers types de fuels lourds. Il se transforme, lors de la combustion en SO2, gaz polluant
notamment responsable des pluies acides. Afin de limiter les impacts environnementaux
des gaz de combustion, les législations actuelles imposent aujourd'hui l'emploi de
combustibles et carburants désulfurés pour les usages privés (voiture, chauffage…).
Le coût des combustibles désulfurés ne permet pas de
les utiliser dans les centrales thermiques de
production d'énergie. Il est alors nécessaire de
"désulfurer" les gaz de combustion. L'un des procédés
actuellement employé utilise le lavage de ces gaz de
combustion par une solution aqueuse de CaCl2,la
formation et la précipitation de gypse (CaSO4.6H2O)
permet d'éliminer le SO2présent.
(CO2,H2O)
Gaz de combustion
(CO2, H2O, SO2…)
Dans un tel procédé, il y a donc transfert de matière
entre la phase gazeuse et la phase liquide. En outre, la
formation de la phase solide (CaSO4.6H2O) implique
une étape germination qui peut jouer un rôle important
sur la cinétique de formation et croissance des
précipités de (CaSO4.6H2O).

2005-06 Bernard Pieraggi 5
Notions élémentaires de cinétique hétérogène
Exemple 3 : Prise des ciments hydrauliques
Les processus physico-chimiques qui interviennent lors de la fabrication et de l'utilisation
des matériaux de construction usuels tels que ciment, plâtre et chaux font intervenir des
processus hétérogènes souvent très complexes.
Ainsi, le mécanisme de durcissement du ciment (prise du ciment) fait intervenir des
réactions chimiques complexes. Les ciments usuels (ciment Portland) contiennent, selon les
propriétés recherchées, des proportions variables de silicate tricalcique 3CaO.SiO2ou
C3Spar abréviation, de silicate dicalcique C2S, d'aluminate tricalcique, 3CaO.Al2O3ou C3A,
d'aluminate monocalcique CaO.Al2O3ou CA et de ferro-aluminate 4CaO.Al2O3.Fe2O3ou
C4AF.
L'hydratation de ces constituants conduit à la formation de silicates, d'aluminates et de
chaux hydraté qui se recombinent et se lient en conférant sa résistance au ciment. Par
exemple, l'aluminate monocalcique CA est impliqué dans les réactions suivantes où tous les
constituants, hormis ceux indiqués comme solides, sont en phase liquide:
CaO.Al2O3(solide) + 4H2O = Ca2+ + 2[Al(OH)4]-
Ca2+ + 2[Al(OH)4]-+ 6H2O = Ca[Al(OH)4][Al(OH)6].6H2O(solide)
2[Al(OH)4]-= 2Al(OH)3+ 2(OH)-
2[Al(OH)4]-+Ca2+ + 2(OH)-+ 3H2O = Ca2[Al(OH)4][Al(OH)6].3H2O(solide)
Ces réactions déterminent la résistance des ciments alumineux lors des premières stades
de la prise.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
1
/
55
100%