les aldéhydes et les cétones

CHM 1302 page 98
V- LES ALDÉHYDES ET LES CÉTONES
5.1. LE GROUPEMENT CARBONYLE
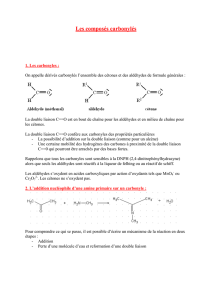
Les aldéhydes et les cétones comportent un groupement carbonyle dans lequel un atome de
carbone hybridé sp2 forme un lien double avec un atome d'oxygène.
CO
H
H
σC-O
πC-O
σC-H
σC-H
n
n
C (2sp2)O (2sp
2)
~120°
1.22 Å
L'atome d'oxygène est plus électronégatif que l'atome de carbone; le groupement carbonyle
est donc polarisé.
C
O
C
O
C
O
δ+
δ-
2.88 D
Électronégativité
(échelle de Pauling)
O3.44
C2.55
Le carbone sp2 d'un groupement carbonyle est conséquemment électrophile et est sujet à
des attaques par les nucléophiles.
C
O
C
O
Nu
Nu
© Richard Giasson

CHM 1302 page 99
5.1. LE GROUPEMENT CARBONYLE (suite)
D'autre part, l'atome d'oxygène du groupement carbonyle possède deux doublets d'électrons
libres et est donc une base.
C
OH
C
O
C
O
HH
La basicité des aldéhydes et cétones en fait des accepteurs de ponts hydrogène;
conséquemment, les aldéhydes et cétones de faibles masses moléculaires sont solubles dans
l'eau.
C
O
HOH
HOHHOH
5.2. NOMENCLATURE DES ALDÉHYDES ET CÉTONES
Les aldéhydes comportent un groupement carbonyle terminal alors que les cétones, un
groupement carbonyle interne.
C
RH
O
C
RR
O
un aldéhyde
(R = H, alkyle, aryle) une cétone
(R, R' ≠ H)
'
© Richard Giasson

CHM 1302 page 100
5.2. NOMENCLATURE DES ALDÉHYDES ET CÉTONES (suite)
NOMENCLATURE DES ALDÉHYDES:
Noms triviaux:
Certains aldéhydes simples sont couramment identifiés par leurs noms triviaux.
H-C-H
O
CH3-C-H
O
formaldéhyde acétaldéhyde
Noms systématiques:
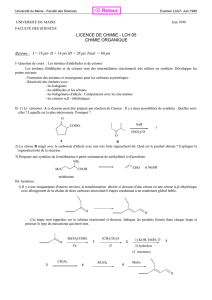
Les noms systématiques (IUPAC) s'obtiennent en considérant les aldéhydes comme des
dérivés des alcanes; la terminaison "e" de ces derniers est alors remplacée par le suffixe "al".
Les aldéhydes deviennent donc des alcanals et le carbone aldéhydique (toujours terminal)
porte d'office le numéro un.
H-C-H
O
CH3-C-H
O
méthanal éthanal
CH3-CH2-C-H
propanal
O
Cl-CH2-CH2-CH2-C-H
4-chlo
r
obutanal
OH
O
7654321
4,6-diméthylheptanal
© Richard Giasson

CHM 1302 page 101
5.2. NOMENCLATURE DES ALDÉHYDES ET CÉTONES (suite)
NOMENCLATURE DES ALDÉHYDES (suite):
Les aldéhydes impossibles à nommer par référence aux alcanes sont plutôt décrits comme
des carbaldéhydes.
CH3CHO
CHO
CHO
OH
OCH3
CHO
1
2
3
4
5
6
cyclooctanecarbaldéhyde 2-méthylcyclopentanecarbaldéhyde-trans
benzènecarbaldéhyde (IUPAC)
benzaldéhyde (ACS) 4-hydroxy-3-méthoxybenzènecarbaldéhyde (IUPAC)
4-hydroxy-3-méthoxybenzaldéhyde (ACS)
NOMENCLATURE DES CÉTONES:
Noms triviaux:
Certaines cétones simples sont couramment identifiées par leurs noms triviaux.
CH3-C-CH3
O
CH3-C
OO
acétone acétophénone benzophénone
© Richard Giasson

CHM 1302 page 102
5.2. NOMENCLATURE DES ALDÉHYDES ET CÉTONES (suite)
NOMENCLATURE DES CÉTONES (suite):
Noms de classe fonctionnelle:
Certaines cétones ont également reçu des noms courants où l'identité des deux groupements
hydrocarbures précède le suffixe "cétone". Cette pratique doit être évitée.
CH3-C-CH3
O
diméthyl cétone
CH3-C-CH=CH2
O
méthyl vinyl cétone
Noms systématiques:
Les noms systématiques s'obtiennent en considérant les cétones comme des dérivés des
alcanes; la terminaison "e" de ces derniers est alors remplacée par le suffixe "one". On
numérote alors la chaîne la plus longue de façon à attribuer le plus petit numéro possible au
carbone du carbonyle, sans égard aux groupes fonctionnels OH, C=C ou C≡C.
CH3-C-CH3
O
CH3-CH2-C-CH3
O
CH3-CH-CH2-C-CH3
OH O
CH3-CH2-CH-C-CH2-CH2-CH3
Br O
1234567
12345
1234
propan-2-one
123
butan-2-one
4-hydroxypentan-2-one
(non pas 2-hydroxypentan-4-one) 3-bromoheptan-4-one
(non pas 5-bromoheptan-4-one)
© Richard Giasson
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
1
/
57
100%