Modifications de chaînes carbonées - Anne CURK

CHIMIE ORGANIQUE 1 : MODIFICATIONS DE CHAINE CARBONEE...............2
I-
ALLONGEMENT DE CHAINE CARBONEE PAR ACTION D'UN C-NUCLEOPHILE SUR UN C-ELECTROPHILE
.............2
1-Les organométalliques : comme doublet nucléophile(révisions bis)..........................2
1-
A
D
EFINITION DES ORGANOMAGNESIENS
2
1-
B
P
ROPRIETES DES ORGANOMAGNESIENS
2
1-
C
S
TRUCTURE DES ORGANOMAGNESIENS
2
1-
D
- R
EACTIVITE DES ORGANOMAGNESIENS
3
2- Les énolates nucléophiles..............................................................................................5
●A
LDOLISATION
: E
TAPE
1 ,
SYNTHESE DES ENOLATES
: 5
●A
LDOLISATION
: E
TAPE
2 , R
EACTIONS NUCLEOPHILES DE L
'
ENOLATE
6
●A
CTION SUR LES DERIVES HALOGENES
12
●A
CTION SUR LES
α-
ENONES
:
LA REACTION DE
M
ICHAËL
12
●R
EACTIVITE ELECTROPHILE D
'
UNE
α-
ENONE
: 12
• (L'équilibre de tautomérie cétoénolique) 14
●C
HOIX DU
H
EN
α 15
●C
ROTONISATION DES ALDOLS OU CETOLS
16
3- Le réactif de Wittig : un ylure de phosphore...............................................................18
3-1O
BTENTION DE L
'
YLURE DE PHOSPHORE A PARTIR D
'
UN DERIVE IODE OU BROME
18
3-2 A
CTION DU REACTIF DE
W
ITTIG SUR LE GROUPE
C=O
DES ALDEHYDES ET CETONES
19
II – MODIFICATIONS DE STRUCTURES CARBONEES PAR MECANISMES CONCERTES.20
1- Les coupures de chaine carbonées (révisions bis)....................................................20
1-
A
- C
LIVAGE DES DIOLS
α
PAR
N
A
IO
4
20
1-
B
- C
LIVAGE DES ALCENES PAR ACTION DE
O
S
O
4
CATALYTIQUE
+ H
2
O + N
A
IO
4
20
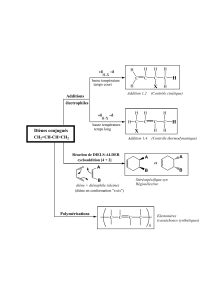
2- La réaction de Diels Alder : obtention de cycles........................................................21
2-
A
- B
ILAN
21
2-
B
- M
ECANISME SELON LEWIS
:
CONCERTE SUPRA
-
SUPRA
21
2-
C
- L
ES INSUFFISANCES DU MODELES DE
L
EWIS
24
3- Métathèse des alcènes..................................................................................................25
3-
A
- L
E MECANISME SIMPLIFIE
26
3-
B
- L
ES
4
TYPES DE BILANS DE METATHESE DES ALCENES
26
3-
C
-
D
ELICATE SELECTIVITE DES REACTIONS DE METATHESE
29
FICHE : OBTENTION DES EPOXYDES (révisions)..........................................31
I - A PARTIR D'UN ALCOOL α HALOGENE OU α HALOGENO ALCOOL ..............................31
II - A PARTIR D'UN ALCENE.....................................................................................................32

2
CHIMIE ORGANIQUE 1 : MODIFICATIONS DE CHAINE CARBONEE
Les méthodes peuvent se répartir en 2 groupes distincts, relatifs au mécanisme des réactions employées :
● L'action d'un carbone nucléophile ( à créer ) sur un carbone électrophile qui conduit systématiquement à
un allongement de la chaîne carbonée
● La recombinaison de chaînes carbonées , spontanée, ou via des catalyseurs bien choisis. Ces méthodes
peuvent reconstruire une molécule, sans obligatoirement allonger la chaîne.
Les méthodes déjà découvertes l'an dernier seront ici rappelées, et complétées conformément au programme.
I- ALLONGEMENT DE CHAINE CARBONEE PAR ACTION D'UN C-NUCLEOPHILE SUR UN C-ELECTROPHILE
Les carbones électrophiles sont légion. Par contre les carbones nucléophiles sont quasi inexistants spontanément,
et toute la difficulté consiste à les obtenir et les maîtriser.
1- Les organométalliques : comme doublet nucléophile (Révisions bis)
1-
A
D
EFINITION DES ORGANOMAGNESIENS
On appelle organométallique tout composé présentant une liaison C – Métal
Exemples : CH
3
– CH
2
– Mg – Br organomagnésien
CH
3
Cu , Li
CH
3
organocuprate lithié
1-
B
P
ROPRIETES DES ORGANOMAGNESIENS
Ils sont tous fabriqués dans un seul but : LE METAL DONNE AU CARBONE LIE UNE POLARITE
δ
-
Le doublet de la liaison C Métal est alors à la fois BASIQUE et NUCLEOPHILE
δ
-
δ
+
Par exemple CH
3
– CH
2
– MgBr / CH
3
– CH
2
– H
Base acide ( l'acide est un alcane : pK
A
≈ 40 )
Pour tous , les acides conjugués étant des alcanes ( ou alcènes , ou alcynes ) , ce sont des BASES TRES
FORTES.
Toutefois, en absence de tout proton acide, ce doublet C – Métal a des PROPIETES NUCLEOPHILES TRES
MARQUEES.
1-
C
S
TRUCTURE DES ORGANOMAGNESIENS
R Mg R Le magnésium présente deux lacunes électroniques .
Dans cet état il est instable et impossible à obtenir.
Il n'existe que stabilisé par solvatation, en présence de DOUBLETS D'ELECTRONS appartenant au solvant :
R Mg X
S
S
où S est un solvant, INDISPENSABLE , donneur de doublet
Ainsi stabilisé, le magnésium est tétravalent (règle de l'octet respectée )
CH
3
– Zn – Br organozincique
Pb-(-CH
2
– CH
3
)
4
Plomb tétra éthyl
CH
3
– Cd – CH
3
Organocadmien
C Métal

3
1-
D
- R
EACTIVITE DES ORGANOMAGNESIENS
Les organomagnésiens permettent d'allonger les chaînes carbonées, par création de liaison C – C en réagissant,
par addition nucléophile, sur les :
♦Aldéhydes et cétones
O
R
1
R
2
δ
+
+
RMgX
R
1
R
2
O
-
R
MgX
+R
1
R
2
OH
R
+
Mg
2+
+
1° étape, milieu anhydre
addition nucléphile
2° étape , hydrolyse acide
H
2
O , H
+
X
-
éther anhydre A/B
(sel)
On a donc créé une liaison C-C au niveau du C électrophile du groupe carbonyle C=O, groupe qui a disparu, se
transformant en alcool, au minimum secondaire.
♦CO
2
O C O
δ
+
+
R MgX
addition
nucléophile
éther anhydre
RO
O
-
MgX
+
RO
OH
+
Mg
2+
+
X
-
H
2
O , H
+
A/B
(sel) acide carboxylique
δ
−
♦Epoxydes
Les époxydes sont des composés très réactifs, et en particulier sensibles aux nucléophiles. En effet ils contiennent
un carbone électrophile ( donc action d'un nucléophile possible ) et insaturé ( par le cyle, donc addition possible )
O
H
δ
+
δ
+
R MgX
addition
nucléophile
OH
R
Mg X
site le moins encombré : REGIOSELECTIVE
attaque anti : STEREOSPECIFIQUE
OH H
R
H
2
O , H
+
A/B
(sel)
+
Mg2+
+
X-
1° étape en solvant anhydre, éther ou THF
2°étape : hydrolyse acide pour libérer un alcool
On a donc allongé la chaîne carbonée :
• Si l'on regarde l'époxyde : on a ajouté un groupe R sur le site le moins encombré de l'époxyde, une
fonction alcool se trouvant sur le C en α.
• Si l'on regarde le dérivé halogéné que l'on avait transformé en organomagnésien : on a rajouté au
minimum 2 carbones : ceux porteurs de la fonction époxyde. La fonction alcool qui est apparue est en β
par rapport au C initialement porteur du dérivé halogéné.

4
♦Esters
R1O
OR2
Ph
O
O
+
R MgX
Ph
O
-
R
O
MgX
Ph
R
O
+
2° action , INEVITABLE
R MgX
H
2
O , H
+
Ph
R
R
OH
+
Mg
2+
+
X
-
δ
+
δ
+
δ
−
add Nu
éther anhydre
Décomposition
spontanée
INEVITABLE
(sel instable)
(sel)
A/B
Ph
O
-
R
R
MgX
fin de la 1° étape, en solvant base de Lewis anhydre
2° étape : hydrolyse acide
+
+
groupe
acyle
groupe partant
alcoolate
et simultanément
H
2
O , H
+
A/B
+ Mg
2+
+ X
-
O
-
MgX
+
O
-
MgX
+
OH
ESTER : δ
+
Nomenclature des esters : Alcanoate d'alkyl
Propanoate de phényle
Benzoate de méthyle
2-chloro éthanoate de 3-méthyl benzyle
L'organomagnésien se fixe
• Une première fois par addition Nucléophile suivie d'élimination. AN/E
• Une deuxième fois par addition nucléophile sur l'aldéhyde ou cétone intermédiaire non isolé : AN
On obtient donc un alcool tertiaire, avec deux groupes R identiques, provenant de 2 molécules d'organomagnésien
fixées.
♥ Un organomagnésien se fixe 2 fois sur un ester.

5
2- Les énolates nucléophiles
●E
TAPE
1 :
SYNTHESE DES ENOLATES
:
Les aldéhydes, les cétones, les esters, présentent une acidité particulière, faible, mais exploitable, du H situé en
α du carbone porteur de la fonction oxo
Ils donnent donc en milieu basique, par équilibre A/B, en enlevant un H éventuellement présent en α du C=O, un
énolate, nucléophile.
R
OHO
-
OH
réf α
R
O
réf α
R
O
-
énolate
forme mésomère retenue
pour l'actvité nucléophile
du CARBONE
forme mésomère qui
donne le NOM à la structure
O N'est PAS nucléophile
(résultat expérimental)
OH O
-
alc
ène
alco
ol
=> énol énolate
Les énols sont instables en général : si une
synthèse les fabrique intermédiairement, ils
se transforment spontanément en cétone
ou aldéhyde, en catalyse acide ou basique.
Nous privilégierons TOUJOURS dans l'écriture de Lewis la forme dont la charge est "posée" sur le carbone : En
effet ,
les énolates sont généralement nucléophiles par l'atome de carbone
. Nous justifierons
ce résultat dans le chapitre "réactivité dans le modèle des orbitales moléculaires", qui permettra d'aller plus loin.
• Choix de la base :
C'est le pK
A
du couple O=C – C – H / O=C – CI
-
qui détermine le choix de la base
Le milieu basique Potasse alcoolique (c'est-à-dire KOH dissous dans l'éthanol) ou ( EtOH / EtO
-
, obtenu par
action du sodium métallique en défaut sur l’éthanol ) sont les plus courants.
Cétone, aldéhyde ou ester : pK
A
≈ 24 (à retenir)
O=C – C – H O=C – C
-
I
H
2
O OH
-
18
14 24
Et – O – H Et – O
-
Les réactions A / B avec la potasse alcoolique ne sont donc pas totales… MAIS, comme l'énolate sera consommé
par son activité nucléophile, les réactions seront déplacées au fur et à mesure, et finalement conduiront sans
problème à l'activité souhaitée. La base est alors un catalyseur.
On peut toutefois obtenir l'énolate par une réaction totale, en choisissant une base de pK
A
plus élevé :
la LDA pK
A
≈ 34
, le n-butyl lithium pK
A
≈ 40
ou des hydrures de lithium ou de potassium pK
A
≈ 40 délicats car H
-
est aussi
nucléophile…
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
1
/
33
100%