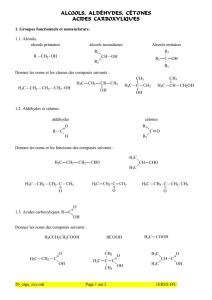
cohrohro + h + hrohhroh

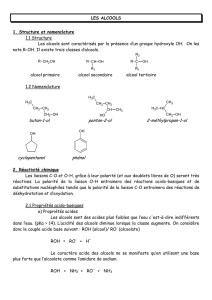
Liaison Carbone Oxygène page 1/4
La liaison Carbone-Oxygène simple
On rencontre cette liaison dans les alcools R-O-H et les étheroxydes R-O-R'.
I Caractéristiques de la liaison C-O dans les alcools
1 Données expérimentales
Les liaisons C-O sont polaires mais moins que les liaisons O-H.
2 Réactivité des alcools: généralités
Voici la cartographie des charges des alcools.
Les réactifs nucléophiles seront attirés par O ou H, (l'alcool se
comporte alors comme un électrophile)
L'alcool est également un nucléophile à cause des doublets non liants
de O, et pourra attaquer un centre électrophile (positif)
II Propriétés acido-basiques des alcools
1 Acidité
Les alcools présentent une acidité très faible pKa >> 14, qui
diminue avec la classe d'alcool, la base associée, l'ion
alcoolate étant déstabilisé par les effets inductifs donneur des groupements.
Préparation des ions alcoolates:
Une base comme la soude est trop faible, on utilise une base plus forte l'ion amidure NH2- (plus exactement
l'amidure de sodium NaNH2): ROH + NH2⊖RO⊖ + NH3
On peut également utiliser une réaction d'oxydoréduction, grâce à un réducteur puissant, le sodium :
ROH + Na RO⊖ + Na⊕+ ½ H2
2 Basicité
De même, les doublets non liants de l'oxygène
donnent une basicité très faible aux alcools.
Là encore la basicité des alcools diminue avec la
classe des alcools. Cette évolution de la basicité
vient de deux effets opposés:
les effets donneurs des groupements qui stabilise l'acide associé (ion alkyloxonium ROH2+) et qui rend donc
l'alcool de plus en plus basique à mesure que le nombre de groupements augmente.
l'effet de stabilisation de ROH2+ par le solvant, affaibli si le nombre de groupements est important, ou si leur
taille est importante. Cet effet rend l'alcool d'autant moins basique que l'alcool est de classe élevée.
C'est le deuxième effet qui l'emporte.
pKb = 16,2
OH
CHH3C
CH3
OH
CH2
H3C
O
H3C
H
H
OH
H3CO
CH2
H3C
H
H
pKb = 17,8
O
CHH3C
CH3H
H
pKb = 16,4
Note: l'effet de solvant intervient aussi pour le caractère acide des alcools, mais va dans le même sens que
l'effet inductif.
OHH
OHC OCH3
HOCH3
H3C
µ = 1,85 D 1,69 D 1,29 D
+
+'
''
')
C-O
O-H
longueur de liaison (nm)
0,143
0,096
Energie de liaison (kJ mol-1)
358
459
+
+
C O H
Nucléophile (centre négatif)
Electrophile
(centre positif)
Base
R O HR O + H
acide très faible base très forte
OH
H3CO
H3COH
C
H3C
H3C
HO
C
H3C
H3C
HO
C
H3C
H3C
H3C
OH
C
H3C
H3C
H3C
pKa = 15,9 pKa = 16,8 pKa = 17,2
+ H R O H
H
R O H
acide très fort
base très faible

Liaison Carbone Oxygène page 2/4
Préparation des ions alkyloxonium:
On utilise des acides forts, par exemple HCl anhydre: ROH + HCl ROH2⊕ + Cl⊖
Note: Il faut que la base conjuguée de l'acide (ici Cl⊖) soit un nucléophile faible, pour éviter une substitution
nucléophile immédiate. En effet le groupement OH2est un
excellent groupe partant, (excellent nucléofuge). On ne
pourra donc pas utiliser HI (I excellent nucléophile).
On peut également utiliser des acides de Lewis,
molécules présentant des atomes possédant un
défaut d'électrons par rapport à la règle de l'octet
(lacune électronique): MgCl2, AlCl3, ZnCl2.
(le groupement HOZnCl est également excellent nucléofuge).
III Propriétés nucléophiles des alcools:préparation des étheroxydes
Les alcools sont également des nucléophiles moyens (doublets non liant), les ions alcoolates sont beaucoup
plus puissants.
Les étheroxydes peuvent être synthétisés suivant la
substitution nucléophile de mécanisme en plusieurs
étapes :
Le bilan est ROH + R'X ROR' + HX
Cette réaction est très lente car ROH est peu
nucléophile.
On lui préfère la synthèse de Williamson dans laquelle on active l'alcool en obtenant l'ion alcoolate (grâce au
sodium par exemple). La substitution qui s'en suit est très facile:
Synthèse de Williamson: (1) ROH + Na RO⊖ + Na⊕ + ½ H2
(2) RO⊖ + R'X ROR' + X⊖
Le bilan des deux phases est : ROH + Na + R'X ROR' + Na⊕+ X⊖+½ H2
La réaction (2) est de type SN2 si R'X est un halogénure primaire ou secondaire.
Si R'X est tertiaire ou encombré, la réaction de substitution (2) est en compétition avec l'élimination d'un H
sur R'X (s'il existe) car la base RO⊖ peut difficilement s'approcher du carbone de R'X.
Voici quelques exemples, où l'on donne également les vitesses relatives vr:
H3COH + CH3I CH3OCH3 + HI vr = 1
H3CO⊖ + CH3I CH3OCH3 + I vr = 2 106 (beaucoup plus rapide)
(H3C)3CO⊖ + CH3I (H3C)3COCH3 + I vr = 3 103 (alcoolate encombré, ici pas d'élimination possible)
Il est possible avec des composés du type halogénoalcool, d'obtenir une synthèse de Williamson
intramoléculaire :
La réaction suivante se
fait facilement en
solution diluée basique.
On obtient l'ion
alcoolate qui évolue par
substitution nucléophile interne (SNi) vers l'étheroxyde cyclique (ici le tétrahydrofuranne ou THF qui est un
solvant organique très utilisé)
O
R
H
H
I+RI + H2O
O
R
H+ Zn ClCl O
R
H
Zn
Cl +Cl
+
O
R
H
R' X O
RR'
H
+X
O
RR'
H
+
O
RR' H
Br OH+OH
CH2CH2
CH2
CH2
O
Br
H2O
O+Br
SNi

Liaison Carbone Oxygène page 3/4
IV Réactions où la liaison C-O se rompt
Cette liaison est solide. Elle ne peut se casser qu'après avoir été activée, en la transformant en ROH2⊕ par
exemple.
1 Synthèse de RX
a Action de HX bilan: ROH + HX RX + H2O
mécanisme: (1) ROH + HX ROH2⊕ + X⊖(réaction acido-basique)
(2) ROH2⊕ + X⊖ RX + H2O (SN1ou SN2)
La deuxième étape est une substitution nucléophile classique de type SN1 et rapide avec les alcools tertiaires
(carbocation stable), et SN2 et plus lente pour les alcools primaires (et secondaires).
La vitesse de réaction est dans l'ordre: I < II < III ce qui se voit dans le test de Lucas des alcools:
test de Lucas: dans un tube à essais contenant le réactif de Lucas (solution de ZnCl2 dans HCl concentré)
on ajoute quelques gouttes d'alcools.
les alcools tertiaires conduisent immédiatement à la formation d'une seconde phase (RCl)
les alcools secondaires conduisent à un trouble lent
les alcools primaires ne réagissent pas sans chauffage.
Au cours de ces réactions, il peut y avoir réarrangements des carbocations intermédiaires vers des carbocations
plus stables. Exemple de l'action de HBr sur un alcool secondaire. On obtient un alkyloxonium qui se
réarrange :
H3CHC HCCH3
CH3
OH2
H3CHC CCH3
CH3
H
Br, SN2HC HC
CH3
Br
H3CCH3
réarrangement
SN1H3CHC CCH3
CH3
H
+ Br H3CHC CCH3
CH3
HBr
minoritaire majoritaire
b Action de réactifs minéraux halogénés (PCl3, PBr3, SOCl2)
Voici les bilans: (X = Cl ou Br) (les mécanismes ne sont pas au programme)
ROH + PX3 3 RX + P(OH)3P(OH)3 = H3PO3 est l'acide phosphoreux.
ROH + SOCl2 RCl + SO2 + HCl
A titre indicatif, voici le mécanisme de la deuxième réaction:
La dernière étape est une substitution nucléophile interne SNi
O
R
H
+ O S Cl
Cl O
R
H
S
O
Cl
Cl
O
RSO
Cl
RCl + O=S=O
HCl SNi

Liaison Carbone Oxygène page 4/4
2 Déshydratation des alcools
a Déshydratation intramoléculaire (élimination)
Synthèse d'alcènes. Bilan:
Condition : il doit exister au moins un H
C'est une élimination classique, elle se fait habituellement en milieu acide sulfurique concentré.
Il y a d'abord une activation du groupement OH : H2SO4 H⊕+⊖OSO3H puis ROH + H⊕ ROH2⊕
Puis il y a élimination assistée par la
présence de la base HSO4⊖(E1 ou E2)
Voici le mécanisme E1:
La réaction est facile (80°C) et de type E1 pour les alcools tertiaires, plus difficile (120 à 150 °C) pour les
alcools secondaires et primaires et de type E2, elle est alors stéréospécifique.
Dans les deux cas de mécanisme, la réaction est régiospécifique (règle de Saytzev).
b Déshydratation intermoléculaire (substitution) Bilan R-OH + ROH R-O-R + H2O
Ce sont les mêmes conditions opératoires: milieu sulfurique, chauffage modéré 100 °C.
Mécanisme:
première étape, activation de l'alcool par protonation de OH : ROH + H⊕ ROH2⊕
deuxième étape, action du nucléophile ROH
il s'agit d'une substitution SN1 ou SN2:
L'ion H⊕ est régénéré et agit comme un catalyseur.
Note: un autre nucléophile du milieu, l'ion hydrogénosulfate HSO4⊖ peut faire la substitution. On obtient alors
l'hydrogénosulfate d'alkyle R-OSO3H. Mais il évolue rapidement par une seconde substitution vers ROR
sous l'action de l'alcool ROH meilleur nucléophile que HSO4⊖.
c Compétition entre élimination inter et intramoléculaire
Les alcools tertiaires sont encombrés, ce sont de mauvais nucléophiles. L'approche difficile de ROH sur
ROH2⊕ rend la formation de l'étheroxyde limitée. Ils conduiront toujours aux alcènes.
Par contre, on obtient un mélange d'alcène et d'étheroxyde dans le cas des alcools primaires et secondaires.
Nous nous retrouvons alors avec la compétition SN/E. On peut privilégier l'élimination en augmentant la
température.
Exemple : l'éthanol CH3CH2OH en milieu sulfurique conduit uniquement à l'éthylène H2C=CH2 à 180 °C, et
uniquement à l'éthoxyéthane CH3CH2OCH2CH3 à 120°C.
C C
OHH
H2SO4
concentré et chaud C C +H2O
C C OH2
H
C C
H
H2OC C
HOSO3H
C C
ROH
OH2
RR O R
H
H2O
R O R + H
1
/
4
100%