SUBSTITUTIONS NUCLEOPHILES ET ELIMINATIONS

SUBSTITUTIONS NUCLEOPHILES ET ELIMINATIONS.....................................2
PREAMBULE : DEFINITIONS.....................................................................................................2
I. REACTIFS TYPIQUES : DERIVES HALOGENES...................................................................2
1-Exemples 2
2-Réactivité 2
II. MECANISMES.........................................................................................................................3
1-Cinétique d’ordre 1 : SN
1
et E
1
3
2- Cinétique d’ordre 2 : SN
2
et E
2
4
3-Tableaux résumés des influences diverses 5
III- GENERALISATION : DIVERSITE DES SUBSTRATS ET DES REACTIFS...........................5
1-Les alcools 5
2- Les amines 9
3- Les hétéroatomes électrophiles 9
REPONSES........................................................................................................10
EXERCICES SN / E 2016 2017 ........................................................................11

2
SUBSTITUTIONS NUCLEOPHILES ET ELIMINATIONS
PREAMBULE : DEFINITIONS
Nucléophile : On appelle nucléophile toute espèce porteur d’un doublet ; σ ou π, libre ou lié, susceptible d’établir
une liaison σ avec un élément chimique, généralement δ
+
ou + ( électrophile ) , différent de H (
fréquemment un C)
.
Electrophile : On appelle électrophile toute espèce présentant un élément différent de H, déficitaire en électron (δ
+
ou +) ET capable d’établir un nouvelle liaison σ avec un doublet d’électron apporté par un nucléophile.
Un réactif porteur d’un Hδ
+
est un acide et non un électrophile, et l’espèce qui établit une nouvelle liaison avec cet
H est appelée une base et non un nucléophile.
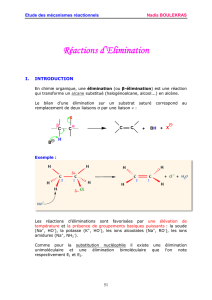
I. REACTIFS TYPIQUES : DERIVES HALOGENES
1-Exemples
Cl
Br IBr N Si
Si
2-Réactivité
•C porteur de l’halogène est un centre
(1)
……………………….
•Coupure de la liaison C-X : coupure
(2)
………………………….., X
-
est un bon
(3)
………………………………
•A priori , les RX seront sensibles aux
(4)
……………………………….., par une attaque sur l’atome de
(5)
……….
Exemples de nucléophiles :
OH C N R OH H O H SH NBr R NH
2
R CH C
O
... RCC
•A posteriori, l’expérience montre que les RX sont aussi sensibles aux BASES, par une attaque sur l’atome d’H
situé sur le C en α du carbone de référence, porteur de l’halogène
Positions acceptables de l’H actif :
Cl
H
HBr I
H
H
Br N Si
Si
H
réf
α
réf
α
α
réf
réf
α
α
α
Exemples de bases :
Hydroxyde Alcoolate alkylure amidure hydrure
OH RO Bu soit NH
2
NH
pK
A
:
(6)
……. ……….. …………. ……….. ……
Remarque : Ordres de grandeur des pK
A
à connaître ( nécessaires toute l’année en chimie organique )

3
II. MECANISMES.
1-Cinétique d’ordre 1 : SN
1
et E
1
Principe : La liaison C-X se rompt spontanément
1° étape , LENTE car rupture de liaison : Obtention d’un carbocation.
Br
+ Br
carbocation PLAN ,
si stabilisé par résonance => réactivité multiple
nécessite d’être relativement stable pour exister=> primaire :NON
2° étape , rapide, car disparition du carbocation : 2 possibilités selon la nature du réactant :
Réactif NUCLEOPHILE : noté Nu
H+ Nu
H
Nu +
H
Nu
RACEMIQUE
Aucune propriété stéréochimique du mécanisme
Il s’est produit une SN
1
Réactif BASE : noté B
-
Recherche d’un H en α du C
+
, acide :
H
H
+B++ HB
MAJ min
Résultat expérimental : l’alcène obtenu est le plus stable .
Cela constitue la règle de Saytseff.
On parle de contrôle THERMODYNAMIQUE lorsque la
nature du produit de réaction est déterminé uniquement
par des considérations de stabilité du produit FINAL,
indépendamment du niveau d’énergie des intermédiaires
de réaction ou états de transition.
Règles de stabilité :
♦tout alcène conjugué est plus stable qu’un alcène non
conjugué.
♦ à défaut de conjugaison, un alcène est d’autant plus
stable qu’il est substitué.
♦ un alcène de configuration E est plus stable qu’un
alcène de configuration Z ( argument stérique)
Il s’est produit une E
1
, réaction sous
contrôle thermodynamique
V = k [R-X] imposée par l’étape lente
SN et E sont en compétition, particulièrement dans le cas d’un réactif à la fois nuclépohile et base , comme OH
-
.
Certains réactifs ne sont pas assez basiques ( Br
-
, RNH
2
) pour envisager une E
Certains réactifs sont beaucoup plus basiques que nucléophiles ( R
2
N
-
, R
3
CO
-
…) => pas de SN
réf
α
1
α
2

4
2- Cinétique d’ordre 2 : SN
2
et E
2
Principe : La liaison C-X ne se rompt que avec assistance, nucléophile, ou basique indirecte.
Réactif NUCLEOPHILE : noté Nu
-
Br
H
+Nu
(R)
Br Nu
H
Nu
H
Br +(S)
♦1 seule étape via l’état de transition mentionné
♦On observe une INVERSION de WALDEN qui
conduit à une inversion de configuration du C* si le
groupe entrant Nu présente la même priorité que le
groupe sortant, dit nucléofuge, Br
-
=>La réaction est STEREOSPECIFIQUE ,
Il s’est produit une SN
2
,
énantiospécifique
Réactif BASE : noté B
-
On observe aussi un contrôle thermodynamique qui
conduit à l’alcène le plus stable, mais en plus :
Pour que la réaction se produise avec assistance, il
est nécessaire ( obligatoire ), que le dérivé halogéné
adopte une conformation telle que C-H et C-X soient
coplanaires et en anti. On parle de conformation
ANTIPERIPLANAIRE :
Cl
HCH
3
H
H
3
C
B
(R) (S)
H
B
Cl
H
H
3
C
CH
3
CH
3
H
3
CH+ Cl
-
BH+(Z)
♦1 seule étape via l’état de transition mentionné
♦L’énantiomère (S) (R) donnerait aussi l’alcène (Z) ,
alors que les diastéréoisomères (R)(R) et (S)(S)
donneraient l’alcène (E) :
=>La réaction est DIASTEREOSPECIFIQUE
Il s’est produit une E
2
,
sous contrôle thermodynamique,
diastéréospécifique
v = k [R-X] . [Nu
-
] ou v = k [R-X] . [ B
-
]
imposée par l’unique étape
La compétition SN / E existe, en ordre 2 comme en ordre 1 .

5
2-Bilans
Bilan SN : RX + Nu
-
RNu + X
-
Bilan E : H-C-C-X + B
-
C=C + X
-
+ HB
3-Tableaux résumés des influences diverses
SN 1
E 1
Alkyle C
+
stable
Halogène I>Br>Cl ( par ordre décroissant de
(7)
……………………..)
Solvant
(8)
………………….., pour stabiliser les C
+
Σ
-
peu actif
Température
(9)
……………………….
(10)
………………….
SN 2
E 2
Alkyle Peu encombré , primaire favorable
Halogène Faible influence
Solvant
(11)
……………………….., pour activer Nu ou B
-
Σ
-
Nucléophile , actif
Base , forte
Température
(12)
………………………….
(13)
…………………………
III- GENERALISATION : DIVERSITE DES SUBSTRATS ET DES REACTIFS
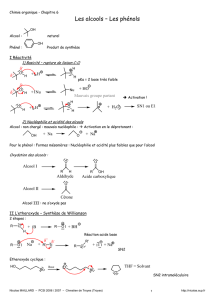
1- Les alcools
♦Les alcools peuvent être nucléophiles par l'atome
(14)
………………………..Ils sont moins nucléophiles que l'eau
car
(15)
…………………………………………………………………………………………………………………………..
Donc les alcools primaires sont
(16)
……………nucléophiles que les secondaires . Les alcools tertiaires quant à eux
sont
(17)
……………………nucléophiles.
Pour augmenter la nucléophilie d'un alcool, on peut le transformer en
(18)
……………………en milieu très
(19)
……………………
pK
A
(20)
…….
Zone de pK
A
des bases nécessaires alcoolate par (R) totale
 6
7
8
9
10
11
12
13
14
15
16
17
6
7
8
9
10
11
12
13
14
15
16
17
1
/
17
100%