Poly Pr Cadiot - polys-ENC

Cirrhose(228) 1
QS 228 - CIRRHOSE ET COMPLICATIONS
- Diagnostiquer une cirrhose
- Identifier les situations d'urgence et planifier leur prise en charge
- Argumenter l'attitude thérapeutique et planifier le suivi du patient
- Décrire les principes de la prise en charge au long cours.
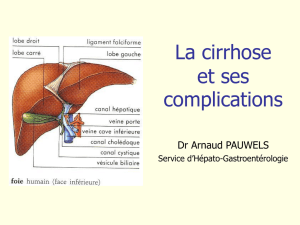
La cirrhose est une maladie chronique du foie caractérisée par une fibrose diffuse mutilante entourant des
nodules de régénération. Elle résulte de lésions hépatocytaires chroniques dont les causes sont multiples.
En France, l'alcool est la principale d'entre elles devant le virus C. L'évolution de la cirrhose se fait en
deux temps : une première période où la cirrhose est non compliquée (cirrhose "compensée") et une
seconde marquée par la survenue de complications : ictère, encéphalopathie, ascite, hémorragie digestive
et carcinome hépato-cellulaire (CHC). La terminologie "cirrhose non compliquée" et "cirrhose
compliquée" doit être préférée à celle de "cirrhose compensée" et "décompensée".
1. Anatomo-pathologie
La cirrhose associe une fibrose diffuse et des nodules de régénération qui sont la conséquence de la
destruction des hépatocytes. Du fait de la fibrose, les nodules de régénération se développent de façon
anarchique, sans connexions normales avec le système vasculaire ou biliaire et ils ne sont donc pas ou peu
fonctionnels.
La taille des nodules permet de distinguer les cirrhoses micro-nodulaires où les nodules sont de petite
taille inférieure à 3 mm, les cirrhoses macro-nodulaires où les nodules sont de taille supérieure à 3 mm et
les cirrhoses mixtes.
La taille du foie permet de distinguer les cirrhoses atrophiques, les cirrhoses hypertrophiques et les
cirrhoses atropho-hypertrophiques. La dimension du foie n'a donc pas de valeur diagnostique.
2 - Epidémiologie
En France, la principale cause de cirrhose est l'alcool (80 à 90 % des cirrhoses chez l'homme et un peu
moins chez la femme). Viennent ensuite le virus C, le virus B (ou BD) et l'hémochromatose génétique.
Les causes plus rares sont la cirrhose biliaire primitive, l'hépatite chronique auto-immune, la cholangite
sclérosante, la thrombose des veines sus-hépatiques, les obstructions prolongées des voies biliaires extra-
hépatiques (cirrhose biliaire secondaire).
Les causes exceptionnelles sont la maladie de Wilson, le déficit en alpha-1-antitrypsine, la protoporphyrie
érythropoïétique.
Des études récentes indiquent que l'obésité souvent associée à un diabète est une cause sous-estimée de
cirrhose par le biais d'une stéato-hépatite.
3 - Diagnostic positif
La cirrhose peut être totalement asymptomatique et découverte fortuitement (cirrhose non compliquée) ou
être révélée par des complications (cirrhose compliquée).
A - Cirrhose non compliquée
Clinique
Le diagnostic de cirrhose peut être évoqué à l'occasion d'un examen clinique, par la palpation d'un foie
dur à bord inférieur tranchant ou bien par la découverte de signes périphériques d'insuffisance hépato-
cellulaire (angiomes stellaires, érythrose palmaire, ongles blancs, subictère, gynécomastie chez l'homme,
troubles des règles chez la femme) et d'hypertension portale (circulation collatérale, ascite,
splénomégalie).

Cirrhose(228) 2
Il peut également être évoqué à l'occasion d'un bilan biologique systématique (ou demandé pour une autre
raison) lorsqu'il existe des anomalies biologiques hépatiques ou bien lors du bilan d'une maladie connue
pour son risque d'évolution vers la cirrhose : éthylisme chronique, portage chronique du virus C ou B
notamment.
Biologie
Les tests hépatiques peuvent être normaux. Ils peuvent mettre en évidence une cytolyse (augmentation des
transaminases), une cholestase (augmentation des GGT et des phosphatases alcalines, l'existence d'une
hyperbilirubinémie à prédominance conjuguée signerait une décompensation ictérique), une insuffisance
hépatocellulaire (baisse du TP, hypoalbuminémie), une augmentation polyclonale des immunoglobulines à
l'électrophorèse des protéines (l'augmentation des Igs A est responsable de l'aspect de bloc beta-gamma).
La macrocytose est habituelle au stade de cirrhose, quelle que soit la cause et n'a plus, à ce stade, de
valeur d'orientation vers un éthylisme chronique. Une neutropénie et une thrombopénie sont possibles en
rapport avec un hypersplénisme.
Un taux élevé d'alpha-foetoprotéine est évocateur de CHC. Un taux supérieur à 500 ng/ml chez un sujet
cirrhotique est pratiquement synonyme de CHC.
Imagerie
L'échographie est systématique. La taille du foie n'a pas de valeur diagnostique (cf § anapath). Le foie
peut apparaitre bosselé (dysmorphie hépatique) du fait des nodules de régénération et de la coexistence de
segments atrophiques et hypertrophiques. Il peut exister des signes d'hypertension portale (veine porte
augmentée de calibre, reperméabillisation de la veine ombilicale, anastomoses portocaves, ascite,
splénomégalie). L'échographie a également pour objectif de dépister une lésion focale (risque de CHC sur
cirrhose) sous forme d'un nodule intraparenchymateux. L'existence d'une thrombose porte doit faire
craindre cette complication.
Un scaner hépatique peut être fait en deuxième intention lorsque le patient est peu échogène ou lorsqu'il
existe un doute sur une lésion intraparenchymateuse.
Endoscopie haute
Une endoscopie haute est indispensable pour préciser l'existence de signes d'hypertension portale : varices
œsophagiennes, varices cardiotubérositaires, muqueuse en mosaïque et ectasies vasculaires antrales
évoquant une gastropathie hypertensive portale. La mise en évidence de varices grade II (varices non
effacées par l'insufflation endoscopique) doit faire envisager la mise en route d'un traitement
prophylactique de l'hémorragie digestive par un beta bloquant non cardiosélectif (avlocardyl).
Biopsie hépatique
La PBH permet de confirmer de façon formelle le diagnostic de cirrhose et quelquefois d'en préciser la
cause. Dans la cirrhose compensée, le bilan de coagulation permet habituellement d'effectuer la biopsie
par voie transpariétale (TP > 50 %, plaquettes > 100.000/mm3). En cas de troubles de la coagulation (ou
en présence d'une ascite - cirrhose compliquée), la PBH doit être réalisée par voie transjugulaire
permettant dans le même temps d'effectuer une prise des pressions sus-hépatiques.
B - Cirrhose compliquée
La cirrhose peut rester asymptomatique pendant de nombreuses années. L'évolution dépend de la cause et
de l'efficacité du traitement proposé (sevrage en cas de cirrhose alcoolique - traitement antiviral en cas de
cirrhose virale - saignées en cas d'hémochromatose génétique). Les complications de la cirrhose résultent
de l'aggravation progressive de l'insuffisance hépatocellulaire et de l'hypertension portale ou du
développement d'un CHC (cf infra). Des facteurs intercurrrents peuvent également intervenir, notamment
des infections bactériennes ou des facteurs médicamenteux. Les complications de la cirrhose sont les
suivantes : l'ascite, le syndrome hépatorénal, l'hémorragie digestive, l'encéphalopathie hépatique, l'ictère.
Ces complications sont souvent associées (par exemple encéphalopathie ou ascite faisant suite à une
hémorragie digestive).
Ascite (cf QS 298 - Ascite)
L'ascite peut rester longtemps latente, détectée uniquement à l'échographie (l'ascite est cliniquement
détectable lorsque son volume atteint 1 litre). Lorsque son apparition ou son aggravation est rapide, on
retrouve souvent un facteur déclenchant comme une maladie infectieuse ou une hémorragie digestive.

Cirrhose(228) 3
L'ascite est souvent précédée d'un météorisme abdominal ("le vent précède la pluie") avec des douleurs
abdominales. L'ensemble peut évoquer un syndrome occlusif. Elle peut aussi se traduire par l'apparition
d'une hernie inguinale, crurale ou ombilicale ou la mauvaise tolérance d'une hernie déjà connue.
Lorsque l'ascite devient cliniquement décelable, elle se traduit à l'examen par une matité des flancs
encadrant la sonorité de la région ombilicale. Le tympanisme périombilical est du aux anses grêles qui
flottent dans le liquide d'ascite. L'épanchement est habituellement mobile (possibilité d'ascite cloisonnée
au cours de l'évolution, notamment en cas de ponctions répétées) et la matité se déplace selon la position
du patient, toujours déclive. Elle donne le signe du flot et le signe du glaçon (cf sémio). Lorsque l'ascite
est abondante, le diagnostic est évident dès l'inspection. Il existe une distension abdominale avec diastasis
des grands droits et éversement de l'ombilic. L'ascite peut s'accompagner d'épanchement pleural le plus
souvent à droite (passage de l'ascite dans la plèvre par un pertuis diaphragmatique) et d'œdèmes des
membres inférieurs. Ces oedèmes peuvent remonter jusqu'à la racine des membres et même infiltrer la
paroi abdominale.
Toute ascite doit être ponctionnée et analysée: taux des protéines (l'ascite de la cirrhose est habituellement
pauvre en protides : moins de 30 g/L, le plus souvent entre 10 et 15 g/L), numération des éléments
(habituellement entre 20 et 100 cellules par mm3, la plupart d'origine mésothéliale), examen cytologique
(recherche de cellules néoplasiques) et bactériologique (examen direct et cultures sur milieux solides
éventuellement complétées par ensemencement sur flacons d'hémocultures au lit du malade), Le taux
d'amylase et la recherche de BK sont demandés si des éléments cliniques ou biologiques orientent vers
une tuberculose (cf QS298 pour le diagnostic différentiel).
Les comlications de l'ascite sont les suivantes :
Dyspnée
L'abondance de l'ascite peut causer une dyspnée par compression diaphragmatique. Chez certains sujets,
la dyspnée est liée à un épanchement pleural dû au passage de l'ascite dans la plèvre du fait de l'existence
d'un pertuis diaphragmatique. Dans ce cas, en raison de la pression négative qui règne dans la cavité
pleurale, l'épanchement pleural peut être abondant alors que l'ascite est minime. Une ponction évacuatrice
rapide de l'ascite et le cas échéant de l'épanchement pleural est le traitement.
Hernie ombilicale et autres, étranglements herniaires et rupture de l'ombilic
L'hyperpression abdominale liée à l'épanchement peut provoquer une hernie ombilicale, voire une hernie
inguinale ou crurale. Une hernie déjà connue peut s'étrangler. Dans les cas extrêmes, la hernie ombilicale
peut devenir volumineuse avec un risque de rupture cutanée qui devient une porte d'entrée pour une
infection du liquide d'ascite. Un traitement chirurgical doit être proposé lorsque la hernie devient
volumineuse et symptomatique sans attendre la rupture.
Infection de l'ascite
Le tableau le plus évocateur d'une ascite infectée est représenté par l'association de douleurs abdominales,
de nausées, de vomissements et à l'examen physique d'une douleur abdominale provoquée ou même d'une
défense ; il y a une élévation de la température, l'ensemble réalisant parfois un tableau pseudo-chirurgical.
Ce tableau typique est cependant rare et une infection du liquide d'ascite doit être suspectée devant les
signes suivants, isolés ou associés: douleurs abdominales spontanées ou décelées par une palpation
légère, nausées ou vomissements, diarrhée récente, modifications thermiques pouvant être soit une
élévation de la température au-dessus de 37,5° soit une diminution au-dessous de 36,5°, accentuation d'un
ictère, apparition d'une encéphalopathie ou d'une insuffisance rénale.
Évoqué, le diagnostic sera confirmé par l'analyse du liquide d'ascite. Le liquide est rarement franchement
purulent avec présence de germes à l'examen direct. La mise en évidence d'un germe à la culture est
inconstante et l'augmentation des polynucléaires au-dessus de 250/mm3 est suffisante pour retenir le
diagnostic d'infection et mettre en route un traitement; dans quelques cas, la culture est positive avec un
nombre de polynucléaires normal; on parle de bactériascite dont la signification est incertaine; il est
recommandé d'effectuer une nouvelle ponction dans les 3 jours; un traitement est parfois mis en route
immédiatement selon le contexte clinique; le taux des protéines est peu modifié.
L'infection spontanée du liquide d'ascite est le plus souvent une infection à bacille Gram-, d'origine
intestinale mais quelquefois une infection hématogène à point de départ urinaire, cutané ou pulmonaire
qu'il faut rechercher. Une ponction antérieure est exceptionnellement en cause (faute d'asepsie).
Troubles hydroélectrolytiques, Syndrome hépatorénal
Une hyponatrémie peut compliquer l'évolution notamment au cours d'un traitement diurétique
avec un régime désodé. L'hyponatrémie inférieure à 130 mmoles/L et l'apparition d'une
insuffisance rénale imposent l'arrêt des diurétiques. Cette insuffisance rénale est fonctionnelle

Cirrhose(228) 4
et est améliorée par le remplissage vasculaire. Le risque est d'évoluer vers le syndrome hépato-
rénal.
Le Syndrome hépato-rénal associe une insuffisance rénale, une oligurie, une hyponatrémie avec une
hyponatriurèse et une ascite irréductible. Ce syndrome est de mauvais pronostic et ne répond pas au
remplissage vasculaire. Il survient à un stade évolué de la cirrhose spontanément ou provoquée par des
complications (hémorragie digestive, infections bactériennes). Des études récentes ont montré qu'un
traitement vasoactif par glypressine et perfusions d'albumine pouvait être efficace dans un certains de cas.
Hémorragies digestives (cf QS 205 hémorragie digestive)
Elles sont le plus souvent dues à la rupture de varices œsophagiennes ou cardio-tubérositaires plus
rarement à la gastropathie hypertensive (gastrite hémorragique). Un ulcère gastro-duodénal associé peut
également être en cause. L'endoscopie haute permet d'identifier la cause de l'hémorragie et éventuellement
d'effectuer un geste d'hémostase endoscopique (sclérothérapie ou ligature de varices oesophagiennes)
Une hémorragie digestive, même jugulée, peut se compliquer d'une ascite, d'une encéphalopathie ou
d'infections bactériennes.
Encéphalopathie
L'encéphalopathie est liée à l'insuffisance hépatique et aux anastomoses porto-caves. Elle semble due à
des substances neurotoxiques d'origine intestinale qui sont insuffisament métabolisées par le foie du fait
de l'insuffisance hépato-cellulaire et qui gagnent donc anormalement la circulation générale. Les
anastomoses porto-caves ont les mêmes conséquences puisqu'elles shuntent le foie.
L'encéphalopathie se traduit initialement par un astérixis isolé, sans trouble de la conscience. La confusion
apparaît dans un deuxième temps. A un stade plus sévère, apparaît un coma sans signe de localisation. Un
signe de Babinski bilatéral est fréquent. Des convulsions peuvent survenir.
L'encéphalopathie hépatique peut compliquer l'évolution spontanée de la cirrhose. Toutefois, il existe
souvent un facteur déclenchant : prise médicamenteuse (sédatifs en particulier benzodiazépines), troubles
hydroélectrolytiques induits ou non par des diurétiques, infections, hémorragie digestive, décompensation
oedémato-ascitique.
Carcinome hépato-cellulaire (cf QS tumeur primitive et secondaire du foie)
L'incidence annuelle du carcinome hépatocellulaire (CHC) en cas de cirrhose est de 2 à 4 %, avec
toutefois des variations selon la cause de la cirrhose. Le risque est plus élevé en cas de cirrhose
alcoolique, virale B ou C et d'hémochromatose génétique.
Une fois la cirrhose constituée, le risque de CHC persiste même si le sevrage alcoolique ou l'éradication
virale ont été obtenus ou si la surcharge en fer a été réduite pas les saignées.
Le CHC peut être révélé par des complications de la cirrhose sous-jacente ou par un syndrome tumoral. Il
peut être dépisté par une surveillance régulière. Les bénéfices d'une surveillance systématique et ses
modalités (échographie tous les 6 mois ou moins - dosages concommittants de l'alphafoetoprotéine) font
actuellement l'objet d'études d'évaluation médico-économique.
4 - Diagnostic étiologique des cirrhoses
A - Cirrhose alcoolique
Les principaux éléments orientant vers une origine alcoolique chez un malade atteint d'une cirrhose sont
l'existence d'une intoxication chronique, actuelle ou ancienne et l'absence d'autres causes.
Les signes cliniques liés à l'alcoolisme sont : pituites matinales, tremblements fins des extrémités, maladie
de Dupuytren, hypertrophie parotidienne, polynévrite.
Biologiquement, l'augmentation des GGT et la macrocytose n'ont plus de valeur d'orientation dès qu'il
existe une cirrhose.
A l'examen anatomo-pathologique, il existe typiquement une cirrhose micro-nodulaire avec stéatose et
présence (inconstante) de corps de Mallory (en sachant que les corps de Mallory ne sont pas
pathognomoniques d'une prise excessive d'alcool).

Cirrhose(228) 5
Au total, il peut exister des éléments d'orientation vers l'origine alcoolique mais il s'agit toujours d'un
diagnostic d'élimination. Il faut aussi souligner la possibilité de cirrhoses d'origine mixte, à la fois
alcoolique et virale B ou C ou bien alcoolique et liée à une hémochromatose génétique.
Traitement : il est indispensable d'obtenir l'arrêt complet et définitif de l'intoxication, le sevrage absolu
permet parfois d'interrompre l'évolution de la maladie. Le risque de CHC persiste toutefois si le sevrage a
été obtenu au stade de cirrhose déjà constituée.
B - Cirrhose vuirale B, BD et C.
Cf QS hépatite virale
Traitement :
1) En cas de cirrhose virale C, un traitement antiviral est possible à un stade non compliqué : la bithérapie
par Interféron et la Ribavirine a une efficacité réduite (mais non nulle) lorsque le stade de cirrhose est
atteint. Le traitement semble réduire le risque de complications et en particulier le risque de CHC, au
moins lorsqu'une éradication virale ou une normalisation des transaminases a été obtenue. Lorsque la
cirrhose est compliquée (ascite, ictère,encéphalopathie), le traitement antiviral est contre-indiqué. Il faut
discuter l'indication d'une transplantation hépatique.
2) En cas de cirrhose virale B ou BD, un traitement par interféron est possible seulement à un stade non
compliqué. En cas de cirrhose sévère ou décompensée, un traitement par lamivudine (Zeffix®) ou
adéfovir (ATU de cohorte) est possible. Ce traitement est aussi proposé chez les patients qui sont en
réplication et en attente d'une transplantation hépatique (le risque d'hépatite sur greffon est majeur si les
patients sont transplantés en phase réplicative et l'évolution peut alors se faire rapidement vers une
cirrhose du greffon; il faut donc stopper la réplication avant transplantation et mettre en route un
traitement prophylactique par Igs antiHBs à fortes doses après la transplantation). Le problème actuel des
traitements prolongés par lamivudine est la sélection de virus mutants.
C - Hémochromatose génétique (cf QS 242)
C'est une affection génétique transmise selon un caractère autosomal récessif qui se caractérise par une
surcharge en fer de nombreux organes et du foie en particulier. Il s'agit d'une maladie fréquente : la
prévalence de l'hémochromatose génétique en France est évaluée à 4/1000. La principale mutation en
France est la mutation C282Y retrouvée à l'état homozygote chez 95 % des sujets atteints
d'hémochromatose génétique. D'autres mutations ont été identifiées ou sont en cours d'identification.
Le diagnostic est facilement évoqué lorsque sont réunis plusieurs signes de la maladie : l’asthénie, la
mélanodermie, une hépatomégalie ferme et régulière, au début sans insuffisance hépato-cellulaire ni HTP;
des manifestations ostéo-articulaires; le diabète qui devient insulino-dépendant au cours de l'évolution
avec les mêmes risques dégénératifs que le diabète commun; l'atteinte cardiaque inconstante : troubles du
rythme, troubles de la conduction auriculo-ventriculaire et au maximum insuffisance cardiaque
congestive.
Actuellement, le diagnostic est posé le plus souvent à un stade plus précoce : enquête familiale, bilan
d'une augmentation des transaminases, découverte d'une anomalie du bilan ferrique lors d'un examen
systématique, voire bilan d'une asthénie ou de douleurs articulaires.
Les tests hépatiques sont normaux au début ; à un stade plus avancé, des anomalies en rapport avec la
cirrhose apparaissent.
Le coefficient de saturation de la sidérophiline est toujours > 50 %. C'est le signe le plus sensible de la
surcharge en fer. Il précède l'élévation de la ferritinémie. Dans ces cas, la présence à l’état homozygote de
la mutation C282Y (située sur le chromosome 6) permet de confirmer le diagnostic chez 95% des sujets
environ.
La ponction biopsie hépatique avec coloration de Perls met en évidence une surcharge ferrique importante
prédominant dans les hépatocytes associée à une fibrose puis à une cirrhose selon le stade évolutif de la
maladie. Le risque évolutif est le carcinome hépatocellulaire.
Le traitement de l’hémochromatose est la diminution de la surcharge ferrique par des saignées de 400 à
500 ml d'abord hebdomadaires jusqu'à normalisation des stock en fer puis espacées à titre préventif. Le
traitement des cas asymptomatiques est le même ; il permet d'éviter le développement de la maladie.
D - Cirrhose biliaire primitive
La "cirrhose" biliaire primitive est une affection caractérisée par une destruction progressive des petits
canaux biliaires intra-hépatiques. Malgré le nom consacré, il n’y a pas de cirrhose au stade initial de la
maladie. Actuellement, le diagnostic est porté de plus en plus fréquemment et de plus en plus tôt au cours
de l'évolution de la maladie en raison de la fréquence des demandes d'examens biologiques.
 6
7
8
6
7
8
1
/
8
100%