Traitements antiangiogéniques au cours des cancers bronchiques M

La Lettre du Pneumologue - Vol. X - n° 5 - septembre-octobre 2007
Mise au point
Mise au point
158
Traitements antiangiogéniques
au cours des cancers bronchiques1
Antiangiogenic treatments in lung cancer
IP P. Saintigny*, R. Etessami*, J.F. Morère*
1. © La Lettre du Cancérologue 2007;3:71-7.
* Service d’oncologie médicale, hôpital Avicenne ; université Paris-XIII, Bobigny.
Abréviations
CBNPC : cancer bronchique non à petites cellules.
VEGF : Vascular Endothelial Growth Factor.
VEGFR : Vascular Endothelial Growth Factor Receptor.
FGF : Fibroblast Growth Factor.
FGFR : Fibroblast Growth Factor Receptor.
PDGFR : Platelet Derived Growth Factor Receptor.
EGFR : Epithelial Growth Factor Receptor.
DMV : densité microvasculaire.
HR : hazard-ratio.
RÉSUMÉ
Comme dans d’autres tumeurs solides, l’angiogenèse tumo-
rale joue un rôle important dans la croissance tumorale et
dans la diffusion hématogène des cancers bronchiques non
à petites cellules. Les facteurs diffusibles proangiogéniques
se lient à des récepteurs spécifiques des cellules endothé-
liales entraînant la fabrication de néovaisseaux tumoraux.
Les traitements antiangiogéniques en cours de développe-
ment sont soit des anticorps monoclonaux (bévacizumab,
anticorps antivascular endothelial growth factor), soit des
inhibiteurs de tyrosine kinase (sunitinib, sorafenib, vande-
tanib). En première ligne métastatique chez des patients
atteints d’un cancer bronchique non à petites cellules non
épidermoïdes, l’adjonction de bévacizumab au carbopla-
tine-paclitaxel améliore la survie globale par rapport à la
chimiothérapie seule, au prix d’une toxicité non négligeable.
Des résultats encourageants ont été obtenus dans des
essais cliniques évaluant des inhibiteurs de tyrosine kinase
possédant des propriétés antiangiogéniques en situation
d’échec à une chimiothérapie de première ou deuxième
ligne, que ce soit en monothérapie ou en association avec
des inhibiteurs du récepteur à l’epithelial growth factor.
Mots-clés : Cancer bronchique non à petites cellules - Can-
cer bronchique à petites cellules - Bévacizumab - Sunitinib -
Sorafenib - Vandetanib - Thalidomide - Angiogenèse tumo-
rale - Traitement antiangiogénique.
SUMMARY
As in other solid tumors, angiogenesis plays a major role
in tumor growth and carcinomatous cells haematogenous
diffusion during non-small-cell lung carcinoma. Diffusible
proangiogenic factors bind to specific receptors of endothe-
lial cells stimulating neoangiogenesis. New antiangiogenic
therapies are monoclonal antibodies (bevacizumab, antibody
directed against vascular endothelial growth factor) or tyro-
sine kinase inhibitors (sunitinib, sorafenib, vandetanib). In
metastatic non-squamous non-small-cell lung carcinomas,
bevacizumab associated to carboplatine-paclitaxel improves
overall survival compared to chemotherapy alone, with an
increased risk of toxicity. Encouraging results were obtained
in clinical trials evaluating tyrosine kinase inhibitors with anti-
angiogenic activity in advance non-small-cell lung carcino-
mas after failure of prior first- or second-line chemotherapy,
both in monotherapy or in association with epithelial growth
factor receptor inhibitors.
Keywords: Non-small-cell lung cancer - Small-cell lung cancer -
Bevacizumab - Sunitinib - Sorafenib - Vandetanib - Thalidomid -
Tumor angiogenesis - Antiangiogenic therapies.
Au cours de l’évolution des tumeurs solides, la dissémi-
nation tumorale se fait par voie vasculaire sanguine,
vasculaire lymphatique, et parfois au travers d’une
cavité ou le long d’une surface (par exemple, dans la cavité
pleurale ou péritonéale). Les mécanismes de dissémination
vasculaire sanguine sont bien connus depuis la description
de l’angiogenèse tumorale, du concept de switch angiogé-
nique et la mise en évidence des premiers facteurs diff usibles
synthétisés par les cellules tumorales ou les cellules du stroma
tumoral et ayant une activité proangiogénique (1).
J. Folkmann fut le premier, dès 1971, à envisager l’utilisation
d’agents antiangiogéniques dans le traitement des cancers (2).
La meilleure compréhension des mécanismes impliqués dans la
diff usion vasculaire sanguine a permis de développer plusieurs
stratégies, dont les cibles sont soit des facteurs diff usibles
sécrétés par les cellules tumorales stimulant la prolifération
des cellules endothéliales (développement d’anticorps comme
le bévacizumab, anticorps anti-vascular endothelial growth
factor A ou VEGF-A), soit des récepteurs membranaires
portant le plus souvent une activité tyrosine kinase et situés à
la surface des membranes cellulaires (développement d’inhibi-
teurs de tyrosine kinase). Ces molécules sont développées soit
en association à la chimiothérapie ou à d’autres thérapie
ciblées, soit en monothérapie. L’anticorps anti-VEGF-A

La Lettre du Pneumologue - Vol. X - n° 5 - septembre-octobre 2007
Mise au point
Mise au point
159
(bévacizumab, utilisé en routine dans les cancers colorectaux
métastatiques et qui devrait l’être rapidement dans les cancers
bronchiques non à petites cellules (CBNPC), en est probable-
ment la forme la plus aboutie (3).
GÉNÉRALITÉS SUR L’ANGIOGENÈSE TUMORALE (1)
En l’absence de nouveaux vaisseaux, une tumeur ne dépasse pas
1 à 1,5 mm de diamètre. En réponse à l’hypoxie, une activation
des cellules endothéliales (switch angiogénique) se produit et est
à l’origine de l’apparition d’une néovascularisation tumorale. On
diff érencie habituellement l’angiogenèse tumorale (ou néoangio-
genèse), qui correspond à la mise en place de nouveaux vaisseaux
tumoraux à partir des vaisseaux préexistants (par ramifi cation,
formation de ponts ou intussusception), de la vasculogenèse
tumorale, qui est la fabrication de nouveaux vaisseaux tumoraux
à partir de précurseurs endothéliaux mobilisés dans la moelle
osseuse. De nombreux facteurs proangiogéniques et antian-
giogéniques ont été identifi és. Le switch angiogénique apparaît
lorsque la balance est en faveur des facteurs proangiogéniques.
Ces derniers sont soit des facteurs diff usibles, soit des récep-
teurs membranaires, et sont exprimés par les cellules tumorales,
les cellules infl ammatoires ou les cellules endothéliales.
Que ce soit dans le cadre de la néoangiogenèse ou de la vascu-
logenèse tumorale, une déstabilisation des vaisseaux préexis-
tants est nécessaire. La sécrétion par les cellules tumorales
et les cellules endothéliales de protéases entraîne la destruc-
tion de la membrane basale et de la matrice extracellulaire.
L’altération des jonctions entre les cellules endothéliales et le
relargage de facteurs proangiogéniques à partir de la matrice
extracellulaire permettent la migration et la prolifération des
cellules endothéliales, formant des ramifi cations puis des
canaux dirigés vers la source des stimuli. La lumière vasculaire
apparaît ensuite grâce à des interactions entre des protéines
de surface cellulaire et la matrice extracellulaire. C’est donc
un dialogue étroit entre les cellules tumorales et les cellules
du stroma (cellules endothéliales, cellules infl ammatoires) qui
permet le développement des vaisseaux tumoraux.
La vascularisation tumorale est hétérogène au sein des
tumeurs, avec des zones plus richement vascularisées (hotspot)
situées en périphérie. Les capillaires néoformés ont une archi-
tecture anormale et chaotique : larges pores et fenestrations
liés à une mauvaise cohésion entre les cellules endothéliales,
absence de membrane basale et modifi cation des péricytes.
Les vaisseaux présentent des modifi cations anarchiques de
leur diamètre, avec un aspect tortueux et ectasique, et parfois
une structure en mosaïque, qui correspond à l’intégration de
cellules tumorales au sein de la paroi vasculaire au même titre
que les cellules endothéliales. Ils fonctionnent également de
façon anormale, avec une augmentation de la perméabilité
capillaire, la présence de fl ux intermittents, voire inversés, la
présence de zones d’hémorragies focales et de communica-
tions artérioveineuses. Ces capillaires se développent au sein
d’un interstitium, lui-même anormal, œdémateux, et dont la
pression est anormalement élevée. Ces caractéristiques très
générales ne doivent pas cacher une grande hétérogénéité
de la vascularisation suivant le type tumoral.
OBJECTIFS ET CIBLES CELLULAIRES
DES TRAITEMENTS ANTIANGIONÉNIQUES
Les thérapeutiques antiangiogéniques ont pour but de réduire
la croissance tumorale et la diff usion métastatique en diminuant
la perfusion tumorale (3-6). Les cibles cellulaires permettant
d’inhiber la vascularisation tumorale sont multiples. Ce sont
principalement les cellules endothéliales, avec pour objectifs :
de prévenir la formation de néovaisseaux sanguins ou
lymphatiques (véritables traitements antiangiogéniques, par
exemple le bévacizumab) ;
de déstabiliser les néovaisseaux formés (ANET pour Anti-
NEovascular erapy) par l’utilisation de vasculotoxiques (VDA
pour Vascular-Disrupting Agents) ou de la chimiothérapie
métronomique ;
ou, au contraire, de permettre leur “normalisation” afi n
d’améliorer la pénétration intratumorale des cytotoxiques selon
le concept de R.K. Jain (7).
Les autres cibles cellulaires sont les cellules musculaires
lisses et les péricytes, avec pour objectif de déstabiliser les
vaisseaux, ainsi que les cellules stromales afi n de diminuer la
pression interstitielle intratumorale, améliorant ainsi la péné-
tration intratumorale d’autres thérapeutiques. L’inhibition du
recrutement des cellules endothéliales progénitrices d’origine
médullaire (autre eff et de la chimiothérapie métronomique) ou
des cellules infl ammatoires proangiogéniques, représente une
autre voie possible, de même que la stimulation de la sécrétion
de facteurs antiangiogéniques par les cellules dendritiques.
RATIONNEL DU DÉVELOPPEMENT DES TRAITEMENTS
ANTIANGIOGÉNIQUES DANS LE CANCER
BRONCHIQUE NON À PETITES CELLULES CBNPC
Le cancer du poumon est l’une des principales causes de décès
par cancer dans les pays développés. En France, le cancer du
poumon a été la cause de 25 799 décès sur 27 500 nouveaux cas
diagnostiqués en 2002 (8). Malgré quelques progrès récents,
le pronostic reste sombre ; les CBNPC, qui représentent 75 à
80 % de l’ensemble des cancers du poumon, ont une survie
globale à 5 ans de l’ordre de 12 à 15 % (9). Depuis 25 ans, les
progrès dans la prise en charge des CBNPC inopérables n’ont
pas permis d’en améliorer le pronostic. Dans ce contexte, les
thérapies ciblées, en particulier antiangiogéniques, représen-
tent un espoir pour les patients.
De nombreuses études ont évalué l’impact de la densité microvas-
culaire (DMV) dans les CBNPC. Les marqueurs utilisés, dont la
spécifi cité pour les cellules endothéliales sanguines a été discutée,
ont été, suivant les études, le facteur VIII, le CD31 ou le CD34.
Une méta-analyse des données de la littérature (32 études ayant

La Lettre du Pneumologue - Vol. X - n° 5 - septembre-octobre 2007
Mise au point
Mise au point
160
inclus 4 399 patients) a montré l’impact péjoratif d’une DMV
élevée, qu’elle ait été évaluée par l’expression du facteur VIII (HR :
1,81 ; IC95 : 1,16-2,84), du CD34 (HR : 1,99 ; IC95 : 1,53-2,58) ou
du CD31 (HR : 1,80 ; IC95 : 1,10-2,96) [10]. Ces résultats ont été
critiqués en raison de l’absence de standardisation des techniques
de comptage de vaisseaux, en particulier dans la sélection des
hotspots qui permettent de déterminer la DMV (11).
Les principaux acteurs de l’angiogenèse tumorale évalués dans
les CBNPC sont le VEGF-A et ses récepteurs, VEGF Receptor-1
(VEGFR-1 ou Flt-1) et VEGFR-2 (Flk-1 ou KDR), et le basic
FGF (bFGF) et son récepteur FGFR-1 (11). Le VEGF-A appar-
tient à la famille du VEGF ; son isoforme la plus puissamment
angiogénique est le VEGF165. Il agit en se liant à la portion extra-
cellulaire de l’un de ses récepteurs tyrosine kinase exprimé à
la surface des cellules endothéliales, entraînant leur dimérisa-
tion et l’activation de leur activité tyrosine kinase. Le VEGF-A
est sécrété par les cellules du stroma, les macrophages et les
cellules tumorales. La production de VEGF-A est principale-
ment stimulée en réponse à l’hypoxie intratumorale. Dans la
majorité des études, l’expression en immunohistochimie du
VEGF-A est associée à un mauvais pronostic et à une DMV
élevée. Dans une méta-analyse publiée en 2002 (15 études
ayant inclus 1 549 patients traités pour un CBNPC), l’expres-
sion du VEGF-A était un facteur pronostique péjoratif, avec
un HR à 1,48 (IC95 : 1,27-1,72) [12]. Des polymorphismes du
gène codant pour le VEGF-A ont été associés à un niveau
d’expression variable de VEGF-A par les cellules tumorales
de CBNPC (13). Leur rôle dans la progression tumorale n’est
pas bien connu. Enfi n, et comme cela a été décrit dans d’autre
tumeurs, les CBNPC expriment fréquemment le VEGFR-1 et
le VEGFR-2, favorisant ainsi la prolifération tumorale par des
boucles de régulation autocrine/paracrine (11).
Le bFGF appartient à la famille du FGF ; il s’agit également d’un
facteur proangiogénique puissant. Son expression a été étudiée
dans les CBNPC ; elle est retrouvée dans 50 à 75 % des cas, et a été
associée à un mauvais pronostic dans certaines études (14, 15).
Un impact pronostique péjoratif du taux sérique ou plasma-
tique des VEGF-A et bFGF a été retrouvé dans certaines séries
de CBNPC (16, 17). Cependant, les résultats sont contradic-
toires dans leur ensemble (18, 19), en raison de l’absence de
standardisation des tests disponibles, des controverses sur la
réalisation du dosage dans le plasma, le sérum ou le sang total,
de la diffi culté de défi nir un seuil qui est très variable d’une
étude à l’autre, et du relargage de VEGF par les plaquettes et les
leucocytes pendant le prélèvement et lors de la manipulation
des échantillons (11). Ce sont probablement certaines de ces
raisons qui expliquent pourquoi les marqueurs biologiques séri-
ques ne permettent pas actuellement, dans la majorité des cas,
de prédire la réponse aux thérapeutiques antiangiogéniques.
INHIBITEURS DE L’ANGIOGENÈSE DANS LES CBNPC
Seules les molécules les plus avancées dans leur développe-
ment clinique seront abordées.
Bévacizumab
Anticorps recombinant humanisé anti-VEGF-A, il représente
actuellement le traitement antiangiogénique le plus abouti
dans la prise en charge des CBNPC. Dans les essais de phase I,
aucune toxicité dose-limitante n’a été observée en monothé-
rapie. En association à la chimiothérapie, il ne semblait pas
exister à ce stade de majoration des toxicités, en particulier de
la toxicité hématologique, liées à la chimiothérapie (20).
Un essai de phase II randomisé incluant 99 patients de
stade IIIB (avec pleurésie métastatique)/IV a évalué l’associa-
tion de bévacizumab (7,5 mg/kg ou 15 mg/kg) à une chimio-
thérapie de type carboplatine (ASC6) + paclitaxel (200 mg/m²)
administrée toutes les 3 semaines (21). Le bras contrôle était
la chimiothérapie seule. Six cycles étaient administrés, et le
bévacizumab était poursuivi jusqu’à progression dans les deux
bras expérimentaux. Les résultats fi gurent dans le tableau I
et montrent la supériorité du bras bévacizumab à la dose de
15 mg/kg. Curieusement, les résultats du bras bévacizumab à
la dose de 7,5 mg/kg semblent sensiblement inférieurs à ceux
du bras contrôle, sans qu’aucune explication claire ait pu être
retrouvée. Des hémorragies graves (hémoptysie ou hématé-
mèse) ont été décrites chez six patients (9 %) et ont entraîné
le décès chez quatre d’entre eux. Ces accidents ont été observés
pour des lésions centrales de plus de 3 cm, dans le groupe
bévacizumab faible dose dans cinq cas, tardivement dans trois
cas (au-delà de 200 jours de traitement), et plus fréquemment
en cas de carcinome épidermoïde (4/13 carcinomes épider-
moïdes versus 2/53 carcinomes non épidermoïdes).
Sur la base de cet essai, l’Eastern Cooperative Oncology Group
(ECOG) a démarré un essai de phase III comparant l’associa-
tion de bévacizumab à la dose de 15 mg/kg à la chimiothé-
rapie (carboplatine ASC6 + paclitaxel 200 mg/m²) administrée
toutes les 3 semaines versus la chimiothérapie seule, chez des
patients ECOG PS 0-1 atteints d’un CBNPC non épidermoïde de
stade IIIB (avec pleurésie métastatique)/IV, excluant les patients
avec des localisations secondaires cérébrales (22). Dans le bras
expérimental, le bévacizumab était poursuivi au-delà de 6 cycles
en monothérapie jusqu’à progression. L’objectif principal était
la survie globale.
Tableau I.
Résultats de l’étude de phase II randomisée évaluant
l’association de bévacizumab (B) [7,5 mg/kg ou 15 mg/kg] à une
chimiothérapie par carboplatine (ASC6) + paclitaxel (200 mg/ m)
[CP] chez des patients atteints d’un cancer bronchique non à petites
cellules de stade IIIB/IV en première ligne.
CP
n = 32
CP + B 7,5 mg/kg
n = 32
CP + B 15 mg/kg
n = 34
Réponse objective 31,3 % 21,9 % 40 %
Temps
jusqu’à progression 5,9 mois 4,1 mois 7 mois
Survie médiane 14,9 mois 11,6 mois 17,7 mois
Taux de survie à 1 an 59 % 41 % 65 %

La Lettre du Pneumologue - Vol. X - n° 5 - septembre-octobre 2007
Mise au point
Mise au point
161
100
80
60
40
0 6 12 18 24
Mois
Hazard-ratio : 0,66
p < 0,001
Survie globale (%)
30 36 42
20
0
100
80
60
40
0 6 12 18 24
Mois
Hazard-ratio : 0,66
p < 0,001
Survie sans progression (%)
30
20
0
Groupe BPC
Groupe PC
A
Groupe BPC
Groupe PC
B
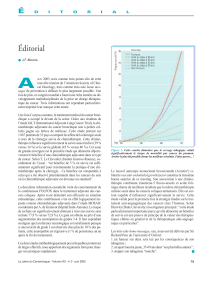
Figure.
Courbes de survie globale et de survie sans progression
de l’essai de phase III comparant, chez plus de 800 patients
atteints d’un cancer bronchique non à petites cellules de
stade IIIB/IV, en première ligne, l’association de bévacizumab
à une chimiothérapie de type carboplatine (ASC6) + paclitaxel
(200 mg/m) [BPC] et la chimiothérapie seule (ECOG) [PC].
Les deux groupes étaient équilibrés, en dehors du sexe, avec
davantage d’hommes dans le groupe chimiothérapie seule
(58 % versus 50 % dans le groupe expérimental, p = 0,03). Le
nombre médian de cycles administrés était de 5 dans le groupe
contrôle et de 7 dans le groupe expérimental. Les résultats
ont été rapportés avec un recul médian de 19 mois. La survie
médiane était de 12,3 mois dans le groupe chimiothérapie +
bévacizumab versus 10,3 mois dans le groupe chimiothérapie
seule (HR : 0,79 ; IC95 : 0,67-0,92 ; p = 0,003). L’évaluation du
critère principal montre donc une supériorité du bras chimio-
thérapie en association au bévacizumab. L’ensemble des résul-
tats fi gurent dans le tableau II et la fi gure.
Cependant, l’administration du bévacizumab était associée à
une majoration de la toxicité : hypertension artérielle, protéi-
nurie, accident hémorragique grave (4,4 % versus 0,7 %, dont
1,9 % versus 0,2 % d’hémoptysies), neutropénie (25,5 % versus
16,8 %), neutropénie fébrile (5,2 % versus 2 %), hyponatrémie,
rashs cutanés et céphalées étaient plus fréquents dans ce
bras (p < 0,05). Il faut noter que l’exclusion des carcinomes
épidermoïdes a permis une nette diminution des hémop-
tysies graves par rapport aux données de la phase II rando-
misée (1,9 % versus 9 %). Le tableau III montre malgré tout
un excès de décès toxiques dans le bras avec bévacizumab,
pour des comorbidités comparables dans les deux groupes
(p = 0,001) ; si la majorité des eff ets indésirables sont apparus
après 3 cycles, la plupart des décès toxiques (5 hémoptysies,
5 neutropénies fébriles, 2 accidents vasculaires, 2 hémorra-
gies digestives et 1 embolie pulmonaire) sont, eux, survenus
au cours des 2 premiers cycles.
Une analyse exploratoire a montré un bénéfi ce de l’adjonc-
tion du bévacizumab à la chimiothérapie dans tous les sous-
groupes considérés en dehors du sexe. Ainsi, seuls les hommes
semblent bénéfi cier de façon statistiquement signifi cative,
Tableau II.
Essai de phase III comparant, chez plus de 800 patients
atteints d’un cancer bronchique non à petites cellules de stade IIIB/
IV, en première ligne, l’association de bévacizumab (15 mg/kg)
à une chimiothérapie de type carboplatine (ASC6) + paclitaxel
(200 mg/m) et la chimiothérapie seule (ECOG).
Carboplatine
+ paclitaxel
n = 433
Carboplatine + paclitaxel
+ bévacizumab (15 mg/kg)
n = 417
Survie globale (médiane) 10,3 mois* 12,3 mois*
Taux de survie à 1 an 44 % 51 %
Taux de survie à 2 ans 15 % 23 %
Survie sans progression
(médiane) 4,5** 6,2**
Réponse objective 15 %*** 35 %***
* HR : 0,79 ; IC95 : 0,67-0,92 ; p = 0,003.
** HR : 0,66 ; IC95 : 0,57-0,77 ; p < 0,001.
*** p < 0,001.
Tableau III.
Causes des décès dans l’essai de phase III comparant,
chez plus de 800 patients atteints d’un cancer bronchique non à
petites cellules de stade IIIB/IV, en première ligne, l’association de
bévacizumab à une chimiothérapie de type carboplatine (ASC6) +
paclitaxel (200 mg/m) et à la chimiothérapie seule.
Variable
Carboplatine +
paclitaxel
n = 440
Carboplatine + paclitaxel +
bévacizumab (15 mg/kg) n
= 427
Décès 344 305
Causes
Cancer bronchique 309 260
Toxicités 2 15
Comorbidités 16 16

La Lettre du Pneumologue - Vol. X - n° 5 - septembre-octobre 2007
Mise au point
Mise au point
162
pour la survie globale, de l’adjonction du bévacizumab. Il est
possible que cette observation soit liée à la prescription plus
fréquente d’une chimiothérapie de deuxième ligne chez les
femmes. Aucune diff érence n’a en tout cas été notée pour ce
qui est de l’utilisation d’inhibiteurs tyrosine kinase du récep-
teur à l’EGF suivant le sexe.
Enfi n, le taux de VEGF circulant à l’initiation du traitement
était comparable dans les 2 groupes, et ce quel que soit le sexe.
Il n’était pas corrélé à la survie globale.
Sunitinib
Le sunitinib est un inhibiteur de l’activité tyrosine kinase de
plusieurs récepteurs transmembranaires : VEGFR-1, VEGFR-2,
mais également Platelet Derived Growth Factor Receptor
(PDGFR)-α, PDGFR-β, Flt-3 et c-KIT. L’intérêt de développer
des inhibiteurs pantyrosine kinase est de pouvoir cibler
plusieurs types cellulaires, en particulier les cellules endo-
théliales (pour les VEGFR) mais également les péricytes qui
expriment les PDGFR et rendent les cellules endothéliales
plus résistantes aux cytotoxiques et aux thérapeutiques anti-
angiogéniques.
Un essai de phase II multicentrique sur 10 sites (États-Unis
et Europe) a inclus 63 patients traités pour un CBNPC de
stade IIIB/IV en échappement après 1 à 2 lignes de chimiothé-
rapie avec ou sans administration d’un inhibiteur de tyrosine
kinase de l’Epithelial Growth Factor Receptor (EGFR), PS 0-1
(23). La dose administrée était de 50 mg/j pendant 4 semaines
consécutives, suivies de 2 semaines de repos. Le traitement était
poursuivi tant qu’il existait un bénéfi ce clinique. La majorité
des patients ont eu une diminution de la taille des lésions cibles.
Le taux de réponse objective était de 9,5 %, le temps médian de
réponse de 12,2 semaines (4,3 à 30,3 semaines) et, chez 42,9 %
des patients, on observait une maladie stable plus de 8 semaines.
Les toxicités étaient les suivantes : asthénie (68 % de grade 1-2,
21 % de grade 3-4), anorexie (40 % de grade 1-2), dyspnée (37 % de
grade 1-2), toux (35 % de grade 1-2), nausées (33 % de grade 1-2,
7 % de grade 3-4), mucite (32 %), dysgueusie (25 % de grade 1-2),
diarrhée (21 % de grade 1-2), vomissements (19 % de grade 1-2,
7 % de grade 3-4), constipation (19 %), hypertension (5 % de
grade 3-4). Il faut noter 3 toxicités de grade 5 : 2 hémoptysies
et une hémorragie cérébrale.
Au total, le sunitinib en monothérapie a une activité promet-
teuse chez les patients ayant un CBNPC prétraités, puisque
environ 50 % des patients ont eu un bénéfi ce clinique. Il a été
généralement bien toléré, et les principaux eff ets indésirables
ont été de grade 1-2 chez des patients en bon état général.
Sorafenib
Le sorafenib (24) est également un inhibiteur de l’activité tyro-
sine kinase de plusieurs récepteurs, la plupart transmembra-
naires : VEGFR-2 et VEGFR-3, PDGFR-β, Flt-3, RAF et c-KIT.
Dans une étude de phase II également présentée à l’ASCO 2006,
les auteurs rapportent les résultats chez 52 patients traités par
une ou 2 lignes (incluant ou non du gefi tinib) pour un CBNPC
métastatique, ne présentant aucun signe de saignement, sans
exclure des patients avec métastases cérébrales asymptoma-
tiques. La dose était de 400 mg x 2/j en continu. Une maladie
stable était observée chez 59 % des patients. Si aucune réponse
objective n’a été notée, une diminution de la taille des lésions
cibles était présente chez 29 % des patients. La survie sans
progression médiane était de 11,9 semaines dans l’ensemble
de la population, et de 23,7 semaines chez les patients stabi-
lisés par le traitement.
Les toxicités les plus fréquentes étaient modérées : diarrhée
(40 % de grade 1-2), syndrome mains-pieds (37 % de grade 1-2,
10 % de grade ≥ 3), asthénie (27 %), nausées (25 %) et hyperten-
sion artérielle (4 % de grade ≥ 3). Un accident hémorragique
mortel a été observé chez un patient traité pour un carcinome
épidermoïde proximal après radiothérapie et 30 jours après
l’arrêt du traitement. La qualité de vie des patients n’a pas été
altérée tout au long du traitement.
Enfi n, les auteurs montrent qu’une concentration de VEGF
plasmatique élevée lors de la mise en route du traitement
est associée à un pronostic péjoratif, au même titre qu’une
absence de diminution franche de sa concentration au cours
du traitement.
Vandetinib
Le vandetinib est un inhibiteur de l’activité tyrosine kinase des
récepteurs transmembranaires VEGFR-2, VEGFR-3, RET et
EGFR. Les études de phase I ont montré qu’il s’agissait d’un
traitement bien toléré à des doses inférieures ou égales à
300 mg/j. Les eff ets indésirables les plus courants sont un rash,
une diarrhée et un allongement du QTc asymptomatique. Les
données de deux phases II sont disponibles (25, 26).
La première a comparé, chez 168 patients ayant reçu 1 ou
2 lignes de chimiothérapie, le gefi tinib 250 mg/j au vandetinib
300 mg/j (partie A). En cas de progression ou de toxicité limi-
tante, un cross-over était prévu (partie B). Les patients avec
métastases cérébrales n’étaient pas exclus, de même que les
carcinomes épidermoïdes, les patients présentant une hémop-
tysie ou une thrombose profonde. L’objectif principal était la
survie sans progression. La proportion de femmes, de non-
fumeurs et d’adénocarcinomes était comparable dans les deux
groupes. L’objectif principal était atteint, puisque la survie
sans progression était de 11 semaines pour le vandetinib et
de 8,1 semaines pour le gefi tinib (HR : 0,69 ; IC95 : 0,5-0,96 ;
p = 0,025). Une réponse objective et un contrôle de la maladie
supérieur à 8 semaines étaient observés respectivement chez
8 % et 45 % des patients sous vandetinib versus 1% et 34% des
patients sous gefi tinib.
Les données disponibles pour la partie B de cet essai montrent
un contrôle de la maladie supérieur à 8 semaines chez 24 %
des patients (7/29) passant du vandetinib au gefi tinib, et
chez 43 % des patients (16/37) passant du gefi tinib au vande-
tinib ; aucune diff érence en survie globale n’a cependant été
observée. Ces données sont diffi ciles à interpréter en raison
des nombreux biais possibles.
Les toxicités étaient principalement de grade 1-2, les diar-
rhées, les céphalées, l’hypertension, l’allongement du QTc et les
>>>
 6
7
6
7
1
/
7
100%