N°2, octobre 2012, la maladie de Horton [PDF

contact
Chers collègues,
Veuillez trouver le
deuxième numéro de
CONTACT, la newsletter
du service de médecine
interne. Dans cette
édition, nous vous
proposons une mise au
point sur la maladie de
Horton. Cette maladie
reste paradoxale :
bien connue en théorie
mais rare en réalité,
souvent typique dans
sa présentation mais
qui fait l’objet de
retard diagnostique….
L’identication précoce
de cette maladie
permet d’éviter
certaines complications
redoutables (cécité,
accidents vasculaires,..).
En cas de suspicion
diagnostique, n’hésitez
pas à nous adresser le
patient en urgence (n°
de référent : 02 47 47 98
88). Le traitement reste
simple dans la plupart
des cas, basée sur une
corticothérapie, avec
une surveillance au long
cours. Bonne lecture !
Bienvenue aux nouveaux
abonnés de CONTACT !
la newsletter du service de médecine interne
>octobre 2012
EDITO
2
LA MALADIE DE HORTON
.../...
La maladie de Horton (MH) ou artérite gigantocellulaire, est une vascularite
de l’adulte de plus de 50 ans. Sa prévalence de l’ordre de 9 cas/100000
personnes en France. Il s’agit d’une panartérite inammatoire à cellules
géantes de topographie segmentaire et focale qui touche les artères de
moyen et de gros calibre, principalement les branches de la carotide externe.
Elle peut atteindre tous les gros troncs artériels. La MH pose des problèmes
diagnostiques.
Quelles sont les principales manifestations cliniques de la maladie
de Horton ?
La présentation clinique classique associe des céphalées d’apparition récente,
une claudication de la mâchoire, une altération de l’état général souvent
fébrile, et à l’examen, une induration d’une artère temporale. Les céphalées
sont généralement temporales et unilatérales. Elles peuvent être associées à
une hyperesthésie du cuir chevelu.
Dans d’autres cas la présentation est plus atypique, reet du caractère
systémique de la maladie. Elle peut se révéler par :
- des signes généraux isolés : èvre ou une perte de poids
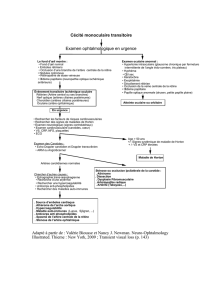
- des signes visuels isolés (ou visuel, baisse d’acuité visuelle brutale, diplopie)
qui font toute la gravité de la maladie
- des signes vasculaires par atteinte de l’aorte ou des gros troncs artériels
(dans 15% des cas, révélés par une claudication de membre, un syndrome de
Raynaud récent, une dissection ou anévrisme de l’aorte) ou à type d’accident
vasculaire cérébral avec atteinte possible des nerfs crâniens (5% des cas).
- des signes respiratoires : toux, dyspnée, douleur thoracique
- des signes dermatologiques : nécrose du cuir chevelu ou de la langue.
Comment diagnostiquer une maladie de Horton ?
Le diagnostic est avant tout clinique. On peut se référer aux critères de
l’American College of Rheumatology (tableau 1). Le signe clinique ayant
la meilleure sensibilité et spécicité est la claudication de la mâchoire
mais présente dans seulement la moitié des cas. La combinaison des
signes, notamment l’association « claudication de la mâchoire + signes
de pseudopolyarthrite rhizomélique + induration temporale » améliore la
spécicité (proche de 100%) tout en diminuant la sensibilité (26%).
Sur le plan biologique, il existe dans la quasi totalité des cas un syndrome
inammatoire : accélération de la VS, augmentation de la CRP. La CRP est
actuellement privilégiée car plus sensible : 5% des maladies de Horton se
présentent en effet avec une VS normale. Seulement 0.8% des malades se
présenteraient avec une VS et une CRP normales.
La conrmation diagnostique doit passer par la réalisation d’une biopsie
d’artère temporale, (tableau 2). La biopsie doit être réalisée de manière
rigoureuse. En raison du caractère segmentaire et focal des lésions, une
longueur minimale de biopsie de 2 cm est recommandée pour analyse en

rédaction : service de médecine interne / maquette : direction de la communication - octobre 2012
plusieurs niveaux de coupes, en privilégiant le côté induré
et/ou douloureux. Le délai de réalisation de la biopsie
doit être court, dès que le diagnostic est évoqué an de
ne pas retarder la mise sous corticoides souvent urgente
du fait des complications vasculaires ophtalmologiques
précoces. La biopsie n’est pas fondamentalement
modiée par une corticothérapie de moins de 10 jours.
Au-delà, l’interprétation devient délicate. Sous réserve
d’une technique de prélèvement et d’analyse rigoureuse,
la biopsie d’artère temporale peut rester négative dans
5 à 10 % des cas, ne permettant pas d’exclure cependant
la maladie si la présentation clinique est fortement
évocatrice. L’attention sera alors portée à l’évolution
sous traitement en reconsidérant le diagnostic en cas
d’évolution atypique.
Quel traitement doit-être instauré lorsque le
diagnostic est suspecté et/ou conrmé ?
Le traitement de référence reste la corticothérapie orale.
La prednisone est privilégiée en raison de sa meilleure
biodisponibilité, en une ou deux prises par jour, selon le
contrôle des symptômes. La posologie initiale est 0.7
mg/kg/j avec un consensus pour majorer la dose à 1 mg/
kg/j en cas de présentation vasculaire sévère notamment
ophtalmologique. Dans ce cas, des bolus intra veineux de
méthylprednisolone sont fréquemment réalisés mais leur
efcacité n’est pas prouvée. Le schéma de décroissance
proposé en France consiste en une diminution rapide
initiale (10% de la dose tous les 7 à 14 jours jusqu’à mi-
dose) puis diminution lente pour une durée totale de
traitement de 12 à 24 mois.
Les traitements associés sont les mesures hygiéno-
diététiques (limiter les apports en sel sans régime sans sel
strict, en graisses et sucres, assurer un apport sufsant
de calcium et de vitamine D). Les biphosphonates
(prévention de l’ostéoporose cortico induite) sont prescrits
systématiquement pour certains ou en cas d’ostéopénie
à l’ostéodensitométrie. L’aspirine à faible dose est
recommandée en raison du risque cardiovasculaire
accru chez les patients atteints de maladie de Horton.
Enn, en cas de complications ischémiques graves,
une héparinothérapie curative courte (7 à 14 jours) est
conseillée. La prescription de potassium et de protecteur
gastrique n’est pas systématique mais se discute en
fonction du terrain.
Existe-t’il des nouveautés thérapeutiques ?
Les corticoïdes étant très efcaces dans la maladie de
Horton, il n’y a d’alternative thérapeutique en première
intention. En cas de corticodépendance majeure,
le méthotrexate peut être prescrit (pas de preuve
formelle en faveur d’une épargne cortisonique). Parmi
les biothérapies, les études avec les anti-TNF α sont
négatives, le TNF ne semblant d’ailleurs pas jouer de
rôle pathogène au cours de la maladie de Horton. Le
tocilizumab (Ac monoclonal anti-Il-6) a été testé sur
quelques malades et pourrait représenter une alternative
thérapeutique intéressante dans la maladie de Horton
mais ces données doivent être conrmées dans des essais
randomisés
Atteinte aortique étendue avec extension aux artères
carotides, vertébrales, iliaques, ect…chez l’une de nos
patientes, atteinte de maladie de Horton
Critères diagnostiques d’artérite gigantocellulaire
1/ âge supérieur ou égal à 50 ans
2/ céphalées localisées et d’apparition récente
3/ sensibilité ou diminution de pulsatilité d’une artère temporale
4/ VS supérieure à 50mm à la première heure
5/ biopsie d’artère révélant une artérite nécrosante avec une prédomi-
nance de cellules mononucléées ou un granulome à cellules géantes
> La présence de 3 critères sur 5 permet d’affirmer le diagnostic avec une sen-
sibilité de 94% et une spécificité de 91% - (American college of rheumatology - 1990)
Critères anatomopathologiques d’artérite gigantocellulaire
- inltrat inammatoire composé de lymphocytes, d’histiocytes, de plasmocytes et de
neutrophiles, présent dans les 3 tuniques artérielles mais prédominant au niveau de la
média. Présence de cellules géantes
- destruction des cellules musculaires lisses de la média
- destruction de la limitante élastique interne
- absence ou discrétion de la brose
1
/
2
100%