PSEUDO POLYARTHRITE RISOMELIQUE ET MALADIE DE HORTON

MALADIE DE HORTON ET PSEUDO POLYARTHRITE RHIZOMELIQUE
La Pseudo Polyarthrite Rhizomélique (PPR.) est un syndrome survenant chez les sujets âgés,
associant une altération de l’état général, des douleurs et un enraidissement des racines des membres et
un syndrome inflammatoire biologique.
Ce syndrome peut-être rencontré de façon autonome ou comme symptôme de au cours de la maladie de
Horton ou artérite giganto-cellulaire qui doit recherchée de principe en raison de ses complications
vasculaires potentielles.
Physiopathologie
La Maladie de Horton est caractérisée par une panartérite giganto-cellulaire segmentaire et focale,
concernant essentiellement les branches de la carotide externe. Elle peut cependant atteindre les
vaisseaux à destinée cérébrale (carotide interne et système vertébro-basillaire) dans leur trajet extra
crânien ainsi que toutes les artères issues de l’aorte, dont les coronaires, les sous clavières, le tronc
cœliaque les mésentériques, les artères rénales, les iliaques communes.... L’évolution de cette atteinte
peut être marquée par la survenue de thromboses artérielles. L’étiopathogénie de cette affection demeure
inconnue. Elle s’observe principalement au sein de la population blanche de l’Europe du Nord. L’incidence
de la maladie est maximale entre 70 et 80 ans et 70% des cas surviennent chez des femmes. Une
prédisposition génétique (commune à la maladie de Horton et à la PPR) est évoquée devant la mise en
évidence de la haute prévalence d’un groupage HLA (HLA DR4) sans que ce marqueur soit suffisament
spécifique pour représenter un argument diagnostique. Des facteurs environnementaux sont actuellement
étudiés, en particulier sur le plan viral ou agents appérentés (Chlamydiae). La probabilité d’une pathogénie
auto-immune est difficile à argumenter devant l’absence de stigmates sériques. Cependant, les caractères
de l’infiltrat observé au sein des artères temporales pathologiques peuvent faire évoquer un processus
immun cellulaire mettant en jeu une synthèse et une libération anormale de cytokines pro-inflammatoires.
Circonstance diagnostique
Les signes de ces affections peuvent s’installer de façon progressive ou ou survenir de façon brutale. Les
signes peuvent évoluer de façon isolée (type fièvre prolongée nue) ou s’associer au sein de syndromes
plus ou moins complets.
Altération de l’état général
L’altération de l’état général associe anorexie, amaigrissement, asthénie et fièvre. Le retentissement sur
l’état général est volontiers massif. La fièvre - présente dans 50 % des cas - est en général un fébricule ou
une fièvre en plateau. Plus rarement toute forme de fièvre prolongée est observable. Il s’y associe en
général des sueurs nocturnes.
Signes rhumatologiques
La pseudo polyarthrite rhizomélique en elle même comporte des arthromyalgies des hanches, des
épaules, du cou avec un enraidissement majeur. L’atteinte est le plus souvent symétrique. Les douleurs
sont à rythme inflammatoire, réveillant le patient en fin de nuit et s’accompagnant d’enraidissement
matinal prolongé, de plus d’une heure.
Douleurs et enraidissements sont responsables d’un handicap fonctionnel parfois majeur dans la vie
quotidienne. Les myalgies des racines sont accentuées par la pression musculaire.
Des arthralgies des grosses et moyennes articulations sont fréquemment rapportées par les patients sans
qu’il existe de véritable arthrite. Des ténosynovites des fléchisseurs des doigts et des oedèmes de la face
dorsale des mains ou des pieds ne sont pas exceptionnels.
Syndrome artériel
Artérite temporale = céphalées et/ou anomalies locales
Artérite temporale = risque ophtalmologique

La maladie de Horton est caractérisée histologiquement par une artérite giganto-cellulaire siégeant le plus
souvent sur les branches de la carotide externe. La traduction clinique la plus fréquente de cette artérite
est une céphalée - présente dans 60 % des cas -, temporale ou fronto-temporale, permanente avec des
paroxysmes, à type de brûlure superficielle, le plus souvent bilatérale, d’intensité variable associée à une
hyperesthésie cutanée en regard des zones douloureuses. Elle peut-être atypique par son siège (facial,
rétro-orbitaire, rétro-auriculaire ou occipitale) par son caractère unilatéral ou par l’absence de signes
locaux d’accompagnement. A l’examen physique les artères temporales sont saillantes sous un
revêtement cutané d’allure inflammatoire, indurées et douloureuse à la palpation avec des pouls
temporaux diminués voire abolis. Plus rarement la maladie de Horton peut se manifester par une
claudication intermittente de la mâchoire (quasi pathognomonique) ou de la langue, des otalgies, des
odontalgies voir une agueusie ou une anosmie.
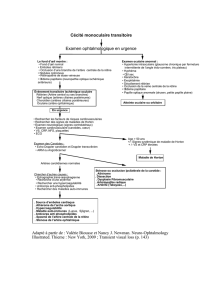
L’atteinte artérielle peut-être révélée par une complication ischémique. Ces complications surviennent
brutalement et sont souvent réversibles. Elles font toute la gravité de la maladie de Horton et explique qu’il
s’agisse d’une urgence diagnostique et thérapeutique. L’atteinte oculaire est la plus grave : elle est parfois
précédée d’une sensation de flou visuel, d’une amaurose transitoire ou d’un épisode de diplopie. Elle
évolue vers une cécité irréversible secondaire à une neuropathie ischémique antérieure aiguë, à une
neuropathie optique rétrobulbaire aiguë ou a une occlusion de l’artère centrale de la rétine.
Artères temporales anormales Complication ischémique locale
(nécrose du scalp)
Autres complications artérielles
D’autres complications ischémiques ont été décrites en relation avec une atteinte des gros tronc artériel
(syndrome de l’arc aortique) : angine de poitrine, infarctus du myocarde, claudication vasculaire des
membres, dissection ou anévrisme aortique, infarctus mésentérique etc.
Complications vasculaires
- sténose artère axillaire - thrombose sous clavière
Atrophie optique

Autres symptômes
La maladie de Horton peut s’accompagner de divers symptômes témoins de localisations diverses (avec
complications locales)
neurologiques de mécanismes discutés : trouble psychiatrique (dépression, confusion, démence) et
ou neurologique central (aphasie, hémiplégie, hémianopsie)
respiratoire sous la forme d’une toux sèche rebelle aux antitussifs
mammaires, ovariennes, prostatiques, osseuses (état lacunaire), hypophysaire (avec pan-
hypoputuitarisme ou sévrétion inappropriée d’ADH) ......
Syndrome inflammatoire biologique
Le syndrome inflammatoire biologique est une des grandes carcatèristiques de la maladie de Horton, la
vitesse de sédimentation étant en général supérieure à 80 mm à la première heure et les diverses
protéines de l’inflammation étant très élevées. Signalons en particulier l’intérêt du dosage du fibrinogène
qui est considéré comme un facteur de risque thrombotique supplémentaire (associé à la thrombocytémie
et au potentiel spontané de l’affection). Ce syndrome inflammatoire retentit sur l’état général avec une
hypoalbuminémie et une anémie inflammatoire. De façon plus anecdotique les fonctions hépatiques sont
altérées dans 25% des cas (élévations des phosphatases alcalines) et on peut observer une hématurie
microscopique. Aucune anomalie immunologique n’est constatée.
Attention :
3 à 5% des patients porteurs d’une maladie de Horton ont une
VS normale ...
Critères de classification de la maladie de Horton
Age de début de la symptomatologie > 50 ans
Céphalées inhabituelle, récente
Anomalie de l’artère temporale à la palpation
VS > 50
Anomalies évocatrices lors de la biopsie de l’artère temporale
Diagnostic retenu lorsque au moins 3 critères sont présents
(sensibilité 93%, spécificité 91 %)

Diagnostic
La biopsie de l’artère temporale apporte la preuve de l’atteinte artérielle montrant une lésion des 3
tuniques. L’artérite étant segmentaire, la biopsie peut-être négative ce qui n’élimine le diagnostic. La
biopsie de l’artère temporale doit être réalisée dès qu’il existe une suspicion de maladie de Horton et elle
ne doit pas retarder l’introduction de la corticothérapie générale puisque les signes histologiques mettent
plusieurs jours, plusieurs semaines pour disparaître. La positivité de la biopsie n’est pas indispensable au
diagnostic. Celui ci repose sur la reconnaissance de 3 critères parmi une liste de 5 définie par l’American
Collège of Rheumatology. Ces critères sont en fait plus des critères de classification que de diagnostic.
Leur crédibilité est donc toujours fonction du contexte bio-clinique.
Diagnostic différentiel
Le diagnostic se discute sur le plan rhumatologique avec :
une polyarthrite rhumatoïde à début rhizomélique (absence de destruction articulaire au cours de
l’évolution de la PPR)
une polymyosite (absence d’élévation des enzymes musculaires ou de syndromes myogènes à
l’électromyographie)
un syndrome RS3PE en cas de PPR avec oedèmes des extrémités et ténosynovite (absence habituelle
de véritable synovite au cours de l’évolution de la PPR)
une poussée de chondro-calcinose articulaire ou de rhumatisme apatitique (absence de liseré calcique
articulaire ou de calcification périarticulaire sur les radiographies)
Parmi les affections extra rhumatologique on évoque essentiellement
les endocrinopathies responsables d’atteintes musculaires telles que les dysthyroïdies
les phénomènes infectieux comme les endocardites subaiguës.
une cause iatrogène : certains anti-hypertenseurs (bêta bloquants, IEC) et certains hypolipémiants
(fibrates et inhibiteurs) pouvant être responsables d’algies rhizoméliques.
et la possibilité d’algies rhizoméliques d’horaire inflammatoire au cours de certaines néoplasies parfois
associées à de véritables PPR paranéoplasiques.
Traitement
La décision de traiter par corticcoïdes, doit-être prise en urgence en raison de la survenue prévisible
d’accidents artériels aiguës au cours de la maldie de Horton. Cette thérapeutique est également utilisée au
cours de la PPR autonome (avec une posologie plus rapidement dégressive).
La corticothérapie est prescrite à la dose d’attaque de 0,5 à 1mg/kg de Prednisone. Le traitement est
efficace en quelques jours sur les douleurs l’état général et l’enraidissement articulaire. Le syndrome
inflammatoire biologique se normalise en quelques semaines. Le traitement d’attaque est maintenu
pendant 4 à 6 semaines puis diminué de façon très progressive. La longueur indispensable du traitement
corticoïde impose la mise en oeuvre de mesures préventives vis à vis du risque minéral osseux.
Les traitements d’appoints comme les anti-paludéens de synthèse n’ont pas fait preuve d’une efficacité
scientifiquement controlée même s’ils sont largements utilisés dans ce cadre de la pathologie
inflammatoire. Les traitements de substitution (Dusulone, méthotrexate) sont en cours de validation.
La surveillance de l’efficacité du traitement repose donc sur la clinique et les paramètres biologiques de
l’inflammation dont la normalité est exigée pour chaque réduction de posologie de corticoïde. De la même
façon ces éléments cliniques et biologiques objectiveront les éventuelles rechutes ou résistance de la
maladie pour adapter la corticothérapie.
L’obtention d’une posologie minimale efficace est le but du traitement en sachant que la durée moyenne
de celui-ci est de 2 ans. A ce terme se posera le problème du sevrage de la corticothérapie.
SEVRAGE DE LA CORTICOTHERAPIE :

Comment éviter le stress des derniers milligrammes ?
Principes
réduction de la dose selon la réponse thérapeutique
pas de prise en compte de la dose totale, de la dose maximale, de la durée de la
corticothérapie
mais tenir compte de la seule posologie résiduelle
avec un test à l’ACTH comme facteur de décision
Pratique
posologie résiduelle supérieure à 10 mg d’équivalent de prednisone : le niveau de
sevrage n’est pas atteint (posologie supra physiologique, les test à l’ACTH est
toujours négatif))
posologie résiduelle inférieure à 5 mg d’équivalent de prednisone : sevrage considéré
comme possible (le test à l’ACTH est toujours positif)
posologie résiduelle comprise entre 5 et 7,5 mg d’équivalent de prednisone,
récupération possible mais non obligatoire (test à l’ACTH pour apprécier la réactivité
résiduelle de la glande).
1
/
5
100%