Lire l'article complet

Interactions médico-chirurgicales
et pneumologie : la 3eJournée
de pathologie thoracique de l’Hôtel-Dieu
Interactions between medical and surgical care in respiratory
medicine : 3rd Respiratory Meeting of “Hôtel-Dieu”
●
C. Lorut*
N
ous remercions à nouveau le comité d’organisa-
tion (Antoine Achkar, Gérard Huchon, Antoine
Rabbat, Jean-François Régnard, Nicolas Roche) et
les partenaires de l’industrie pharmaceutique pour cette 3eJour-
née de pathologie thoracique de l’Hôtel-Dieu. Les thèmes abor-
dés étaient répartis en quatre sessions :
– manifestations respiratoires du reflux gastro-œsophagien (phy-
siopathologie et méthodes diagnostiques, manifestations respi-
ratoires, traitement chirurgical) ;
– pneumopathies interstitielles diffuses chroniques non infec-
tieuses (classification, traitement médical, place de la biopsie
chirurgicale, transplantation) ;
– chirurgie des métastases (processus métastatique, indications
et résultats, intérêt de l’expression du Thyroid Transcription
Factor-1 [TTF1]) ;
– actualité en pathologie thoracique (antibioprophylaxie en chi-
rurgie thoracique, facteurs de gravité des pneumopathies post-
opératoires, paralysies récurrentielles).
PHYSIOPATHOLOGIE DU REFLUX GASTRO-ŒSOPHAGIEN
ET MÉTHODES D’EXPLORATION
d’après le résumé de M. Gaudric
(service d’hépato-gastro-entérologie, hôpital Cochin, Paris)
Le reflux gastro-œsophagien (RGO) est un phénomène phy-
siologique. Ses manifestations cliniques sont fréquentes et sou-
vent banales, et seuls quelques patients consultent pour des
symptômes pouvant être rapportés à un reflux pathologique. Le
sphincter inférieur de l’œsophage (SIO) représente la princi-
pale barrière contre le reflux. C’est une zone de haute pression
permanente qui ne se relâche que brièvement (< 10 secondes)
lors de la déglutition, permettant le passage du bol alimentaire
poussé par une onde de contraction œsophagienne vers la
lumière gastrique.
Le reflux peut survenir dans trois circonstances :
– en cas de relaxation transitoire et spontanée de l’œsophage
(90 à 100 % des reflux chez le sujet normal et deux tiers des reflux
pathologiques) ;
– en cas d’hypotonie ou incompétence du SIO, plus fré-
quente lorsqu’il existe une œsophagite peptique associée.
Il s’agit là de la cause du reflux chez les patients atteints de
sclérodermie ;
– en cas de perte de l’anatomie normale de la jonction œsogas-
trique : la hernie hiatale favorise le reflux par des relaxations
spontanées du SIO plus fréquentes, la diminution du tonus du
SIO par l’absence de pince diaphragmatique et la présence de
liquide acide de stase.
Les principales méthodes d’exploration sont :
– la fibroscopie (dont la sensibilité est faible, mais qui est
utile pour détecter les complications du reflux : œsophagite et
sténose peptiques, endobrachyœsophage ou œsophage de
Barrett) ;
– la pHmétrie des 24 heures (qui est la méthode de référence).
On considère qu’il y a un reflux acide lorsque le pH chute au-
COMPTE-RENDU DE CONGRÈS
203
La Lettre du Pneumologue - Volume VI - no5 - septembre-octobre 2003
* Service de pneumologie et réanimation, hôpital de l’Hôtel-Dieu, Paris.
Thèmes pour cette année 2003 : Manifestations respiratoires du reflux gastro-œsophagien - Pneumopathies interstitielles
diffuses chroniques non infectieuses - Chirurgie des métastases pulmonaires - Actualités en pathologie thoracique.
Topics: Respiratory manifestations of gastro-oesophagal reflux - Non infectious diffuses interstitial pneumoniae - Surgery
of pulmonary metastasis - Update in respiratory medicine.
*PNEUMO 5/2003 20/11/03 11:25 Page 203

dessous de 4 pendant au moins 10 secondes. La manométrie, le
transit œsophagien gastroduodénal et la scintigraphie gastro-œso-
phagienne ont une sensibilité et une spécificité faibles et sont en
pratique peu utilisés. Certains auteurs préconisent également un
test thérapeutique aux inhibiteurs de la pompe à protons à forte
dose en évaluant son impact sur les symptômes du patient.
REFLUX GASTRO-ŒSOPHAGIEN,
ASTHME ET AUTRES MANIFESTATIONS RESPIRATOIRES
d’après le résumé de N. Roche
(service de pneumologie et réanimation, Hôtel-Dieu, Paris)
Plusieurs arguments épidémiologiques, physiopathologiques
et cliniques suggèrent que l’asthme et le RGO sont associés.
Cependant, une grande partie des études sur le lien épidémio-
logique entre RGO et asthme souffrent de biais de sélection :
il s’agit en effet de travaux rétrospectifs ou transversaux por-
tant sur des malades recrutés en gastroentérologie dans des
centres spécialisés. La prévalence de symptômes de RGO dans
ces études est d’environ 70 %, contre 50 % dans des groupes
contrôles non asthmatiques. Chez des asthmatiques non sélec-
tionnés, cette prévalence diminue tout en étant très variable
d’une étude à l’autre : 24 à 63 %. De telles variations sont lar-
gement liées aux différences de critères requis pour parler de
symptomatologie de RGO. La prévalence de l’asthme chez des
malades porteurs d’un RGO n’a fait l’objet que de peu d’études.
Dans l’une d’elles (étude cas-témoins), le risque relatif d’asthme
en cas de RGO était de 1,51 (intervalle de confiance à 95 % :
1,43-1,59). Chez les asthmatiques porteurs d’un RGO, la pré-
valence des symptômes nocturnes (toux, sibilances) est plus
élevée que chez ceux qui n’en souffrent pas. Les trois méca-
nismes par lesquels le RGO peut aggraver l’asthme sont : le
réflexe œsobronchique, qui est controversé, la majoration de
l’hyperréactivité bronchique (HRB) et les microaspirations
(rares au cours du RGO). Les résultats d’une revue systéma-
tique du groupe Cochrane sur les essais randomisés évaluant
le traitement du RGO sont décevants. En effet, les résultats
sont variables d’une étude à l’autre et tous les travaux s’accor-
dent à ne pas trouver de bénéfice spirométrique. Cependant,
dans l’ensemble, environ 60 % des asthmatiques dont le RGO
est traité présentent une amélioration d’au moins un critère
d’évaluation symptomatique de l’asthme. Enfin, plusieurs
limites des études disponibles peuvent être avancées pour expli-
quer les résultats négatifs (exemple : durées de traitement trop
courtes, doses insuffisantes, etc.). En pratique, chez l’asthma-
tique bien contrôlé par le traitement de fond, il semble raison-
nable de prendre en charge le RGO comme dans le cas gé-
néral. Lorsque l’asthme n’est pas correctement contrôlé ou
ne l’est qu’au prix d’un traitement de fond lourd, un RGO
doit être recherché et traité selon la stratégie exposée dans la
figure 1. L’absence d’amélioration doit bien sûr conduire à
rechercher d’autres facteurs aggravants non contrôlés, mais
aussi à vérifier par pHmétrie que le traitement du RGO est suf-
fisant. Enfin, chez les asthmatiques corticodépendants porteurs
d’un RGO récidivant à l’arrêt du traitement médical, il paraît
licite de discuter d’une option chirurgicale. Les autres affec-
COMPTE-RENDU DE CONGRÈS
204
La Lettre du Pneumologue - Volume VI - no5 - septembre-octobre 2003
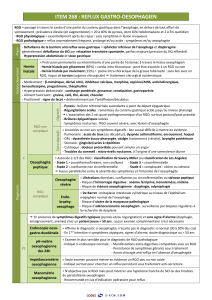
Figure 1. Diagnostic et traitement du RGO de l’adulte, selon la sévérité de l’asthme.
ASTHME SÉVÈRE OU DIFFICILE
Non
pHmétrie sans traitement
Non Stop
Si RGO : traiter à double dose
d’emblée ou en adaptant
les doses selon les symptômes
d’asthme et, en cas d’inefficacité,
selon la pHmétrie
sous traitement
Réponse clinique
Traitement d’entretien
Rechercher le traitement minimal
efficace du RGO
Absence de réponse clinique
pHmétrie sans traitement
Si RGO : majorer son traitement et adapter
les doses par pHmétrie sous traitement
Oui
Test thérapeutique (3 mois) (efficacité jugée sur signes digestifs et asthme)
Symptômes évocateurs de RGO ?
Oui Non
Oui
Symptômes évocateurs de RGO ?
*PNEUMO 5/2003 20/11/03 11:25 Page 204

tions respiratoires dont l’association au RGO est bien docu-
mentée sont la toux chronique et les affections ORL s’accom-
pagnant d’une rhinorrhée postérieure.
INDICATIONS ET RÉSULTATS À LONG TERME
DE LA CHIRURGIE ANTIREFLUX SOUS CŒLIOSCOPIE
SUR LES MANIFESTATIONS RESPIRATOIRES (MR)
ASSOCIÉES AU RGO
d’après le résumé de P. Magdeleinat 1, 3,
F.C. Desmaizières 2, X. de Lavernette 2, C. Genety 2,
A. Petigny 2, M. Duche 4, P. Charvolin 3, J.M. Herlin 1, 3
(1 service de chirurgie thoracique, Hôtel-Dieu,
2 hôpital de Paray-le-Monial, 3 CMC de Creil,
4cabinet de gastroentérologie et d’explorations fonctionnelles
digestives, Compiègne)
Le but de ce travail rétrospectif a été de tenter de préciser les
indications et les résultats de la chirurgie antireflux à long terme.
Quatre cent dix-huit malades consécutifs ont été opérés par fun-
doplicature sous cœlioscopie pour un RGO. Soixante-trois
(15 %) avaient des MR associées à un RGO et ont été retenus
pour notre étude. Quatre-vingt-douze pour cent des patients
avaient une symptomatologie digestive de RGO. Tous ont eu
une fibroscopie digestive haute préopératoire, retrouvant, dans
90 % des cas, une hernie hiatale. Quarante-huit pour cent avaient
une œsophagite préopératoire. Tous avaient au moins un symp-
tôme respiratoire (majoritairement toux chronique ou asthme).
Quatre-vingt-quinze pour cent des patients ont eu des suites
opératoires simples, avec une durée moyenne d’hospitalisation
de 4,7 jours (2 à 8 jours). La mortalité postopératoire a été nulle.
Quatre-vingt-douze pour cent des patients ont été guéris de leur
RGO. La probabilité de guérison du RGO, calculée selon la
méthode de Kaplan-Meier, était de 94 % à 60 mois et de 85 %
à 90 et 120 mois. Soixante-quinze pour cent des patients ont été
guéris ou ont vu leurs symptômes respiratoires s’améliorer. Les
probabilités de guérison ou d’amélioration des symptômes res-
piratoires pour les 63 malades étaient de 77 % à 60 mois, de
68 % à 90 mois et de 56 % à 120 mois. Les critères significati-
vement prédictifs de l’échec de la chirurgie antireflux ont été
la survenue des symptômes respiratoires avant les symptômes
digestifs, le type de MR, le caractère cliniquement atypique des
MR avec un test aux antisécrétoires négatif. L’analyse multi-
factorielle selon la méthode de Cox a retenu deux facteurs pro-
nostiques indépendants : la chronologie des manifestations res-
piratoires et digestives (MD), avec un risque de récidive
supérieur pour les malades ayant des MR précédant les MD
(risque relatif de 3,56 ; IC95 % : 1,14 à 11,11 ; p = 0,03), et le
type des MR, avec un risque de récidive significativement supé-
rieur pour les malades ayant des MR atypiques avec un test aux
antisécrétoires négatif (risque relatif de 6,63 ; IC95 % : 2,20 à
20,1 ; p = 0,0008).
En conclusion, le traitement chirurgical des MR associées au
RGO concerne des malades sélectionnés au terme d’un bilan
établissant le lien entre RGO et MR et reposant essentiellement
sur l’analyse sémiologique des MR et la réponse au test aux
antisécrétoires.
CLASSIFICATION ANATOMOPATHOLOGIQUE
ET RADIOLOGIQUE DES PNEUMOPATHIES INTERSTITIELLES
DIFFUSES CHRONIQUES NON INFECTIEUSES
d’après les résumés de T. Molina 1et M.W. Brauner 2
(1service central d’anatomie et de cytologie pathologiques
Jacques-Delarue, Hôtel-Dieu, Paris,
2service de radiologie, hôpital Avicenne, Bobigny)
Un consensus multidisciplinaire international regroupant des
experts pneumologues, radiologues et pathologistes a proposé
une classification des pneumopathies interstitielles idiopa-
thiques en sept cadres nosologiques (tableau I). L’analyse his-
topathologique des lésions élémentaires du parenchyme pul-
monaire permettra, dans la majorité des cas, de stratifier des
patients dans différents groupes. Chaque groupe lésionnel doit
nécessiter une recherche étiologique large avant d’envisager
une forme idiopathique.
QUAND ET COMMENT BIOPSIER
UNE PNEUMOPATHIE INTERSTITIELLE CHRONIQUE ?
d’après le résumé de Y. Sibille (service de pneumologie,
université catholique de Louvain, Belgique)
La morbidité et la mortalité de la biopsie pulmonaire chirurgi-
cale sont faibles (< 5 %). Toutefois, avant de réaliser une biop-
sie pulmonaire, il est sans doute utile de rappeler l’importance
des données anamnestiques et cliniques qui accompagnent un
tableau de pneumopathie interstitielle diffuse chronique (habi-
tudes tabagiques, exposition aux allergènes, etc.). La biologie
ainsi que l’imagerie peuvent également orienter le diagnostic
(exemple : pneumonie aiguë à éosinophiles, histiocytose X,
etc.). Le débat sur la place des biopsies pulmonaires dites
“chirurgicales” concerne essentiellement les fibroses pulmo-
naires dites “idiopathiques”. L’objectif, suivant les recomman-
dations de l’ERS et de l’ATS, est alors d’établir le diagnostic
de fibrose pulmonaire idiopathique, et en particulier de préci-
ser à quelle sous-classe elle doit être rattachée (UIP, RBILD,
AIP et NSIP). L’enjeu sera particulièrement important, non seu-
lement pour définir la stratégie thérapeutique, mais aussi pour
établir un pronostic de la maladie. La biopsie permet en outre
d’écarter formellement les causes infectieuses, néoplasiques,
voire cardiaques, parfois méconnues malgré une démarche dia-
gnostique appropriée.
QU’ATTENDRE DU TRAITEMENT MÉDICAL
DES PNEUMOPATHIES INTERSTITIELLES CHRONIQUES
NON INFECTIEUSES ?
d’après le résumé de G. Huchon
(service de pneumologie et réanimation, Hôtel-Dieu, Paris)
Les pneumopathies interstitielles chroniques non infectieuses
recouvrent une multitude d’entités, dont certaines ont une cause
identifiée pouvant mener éventuellement à des traitements
ciblés. Lorsque la cause reste indéterminée, les thérapeutiques
à mettre en œuvre dépendent du diagnostic retenu et peuvent
aller de l’abstention avec surveillance à des thérapeutiques beau-
205
La Lettre du Pneumologue - Volume VI - no5 - septembre-octobre 2003
*PNEUMO 5/2003 20/11/03 11:25 Page 205

coup plus agressives, notamment dans le cas des pneumopa-
thies interstitielles fibrosantes idiopathiques. Le traitement est
largement fondé sur l’idée que l’inflammation est en cause. Cela
ouvre la voie à des perspectives thérapeutiques qui restent limi-
tées et font appel aux anti-inflammatoires (corticostéroïdes,
agents cytotoxiques immunosuppresseurs et agents antifibro-
tiques), ainsi qu’à des traitements symptomatiques comme
l’oxygénothérapie et à la transplantation pulmonaire. Devant le
COMPTE-RENDU DE CONGRÈS
206
La Lettre du Pneumologue - Volume VI - no5 - septembre-octobre 2003
Tableau I. Classification clinique, histologique et radiologique des pneumopathies interstitielles idiopathiques.
Diagnostic
clinico-radiologico-pathologique et étiologies
Fibrose pulmonaire idiopathique.
Alvéolite fibrosante cryptogénétique :
Étiologies possibles : collagénoses, pneumopathies
d’hypersensibilité chronique, pneumopathies
médicamenteuses, asbestoses
Pneumopathie interstitielle non spécifique (NSIP),
entité provisoire :
Étiologies possibles : collagénoses, pneumopathies
d’hypersensibilité, médicamenteuses, infections,
déficits immunitaires (VIH)
Pneumopathie organisée (OP) :
Étiologies possibles :
dommages alvéolaires diffus, des infections,
des processus obstructifs, inhalations de toxiques,
prises médicamenteuses, collagénoses,
pneumopathies d’hypersensibilité, poumons
éosinophiles et dans tout processus de réparation
Dommage alvéolaire diffus (DAD) :
Étiologies possibles : infections, collagénoses,
pneumopathies médicamenteuses, inhalations
toxiques, états de choc, traumatismes
Bronchiolite respiratoire (RB) :
Se voit surtout chez les patients tabagiques
Pneumopathie interstitielle desquamative (DIP) :
Étiologies possibles : surtout chez les tabagiques.
Se voient aussi les UIP, NSIP, RB,
poumons éosinophiles, hémosidéroses
Pneumopathie interstitielle lymphoïde (LIP) :
Étiologies possibles :
fréquemment associées à des maladies de système
(Sjögren, etc.), des hypogammaglobulinémies,
ou des déficits immunitaires (VIH, etc.).
Se voit aussi au cours d’infections (pneumocystose),
d’exposition toxique ou médicamenteuse
ou de maladies auto-immunes
(anémie hémolytique, etc.)
Aspect histologique
Pneumopathie interstitielle usuelle (UIP) :
– fibrose dense mutilante avec, souvent,
rayon de miel ;
– présence de foci fibroblastiques ;
– atteinte hétérogène du parenchyme pulmonaire ;
– fréquente prédominance sous-pleurale
et paraseptale des lésions
Pneumopathie interstitielle non spécifique (NSIP) :
– architecture pulmonaire relativement préservée ;
– fibrose habituellement modérée
de répartition homogène ;
– infiltrat inflammatoire interstitiel modéré
Pneumopathie organisée cryptogénétique (COP) :
– fibrose intraluminale des voies aériennes distales ;
– distribution hétérogène des lésions ;
– lésions de même âge ;
– préservation de l’architecture pulmonaire ;
– infiltrat interstitiel inflammatoire chronique
modéré
Pneumopathie interstitielle aiguë (AIP) :
– distribution diffuse ;
– lésions d’âge comparable ;
– fibrose organisée des cloisons interalvéolaires,
habituellement diffuse ;
– fibrose intraluminale des voies aériennes distales ;
– membranes hyalines
Pneumopathie interstitielle associée
à une bronchiolite respiratoire (RB-ILD) :
– légère fibrose et inflammation chronique
bronchiolaire ;
– accumulation de macrophages alvéolaires
pigmentés, empoussiérés ;
– topographie bronchiolocentrique
Pneumopathie interstitielle desquamative (DIP) :
– atteinte uniforme du parenchyme pulmonaire ;
– accumulation prédominante de macrophages
alvéolaires ;
– discrète fibrose des cloisons interalvéolaires ;
– infiltrat inflammatoire chronique modéré
Pneumopathie interstitielle lymphoïde (LIP) :
– atteinte uniforme du parenchyme pulmonaire ;
– atteinte prédominante des cloisons
interalvéolaires ;
– infiltrat dense composé en majorité
de lymphocytes T, plasmocytes et macrophages,
hyperplasie lymphoïde folliculaire
Aspect radiologique
• Réticulations fines intralobulaires (80 %)
• Destructions en rayon de miel (70 %)
à prédominance périphérique et basale
• Bronchectasies par traction (50 %)
• Hyperdensités en verre dépoli (au second plan)
• Hyperdensités en verre dépoli,
le plus souvent bilatérales et symétriques
et à prédominance sous-pleurale
• Épaississements péribronchovasculaires
• Condensations alvéolaires basales
et sous-pleurales
• Opacités réticulées irrégulières
• Destructions en rayon de miel
• Opacités alvéolaires (90 %)
• Topographie le plus souvent périphérique
et inférieure mais parfois péribronchique
• Pouvant être migratrices
• Contenant souvent un bronchogramme aérique
• Hyperdensités en verre dépoli, souvent étendues,
bilatérales et hétérogènes
• Condensations alvéolaires à prédominance
déclive
• Épaississement des parois bronchiques
• Micronodules centrolobulaires mal définis
et de faible densité
• Petits foyers d’hyperdensité sen verre dépoli
prédominant dans les territoires supérieurs
dans 50 % des cas
• Hyperdensités en verre dépoli constantes
et souvent étendues
• Prédominance inférieure (75 %)
et périphérique (60 %)
• Dans 50 % des cas, discrets signes de fibrose
• Hyperdensités en verre dépoli (près de 100 %)
• Souvent associée à des opacités réticulées (50 %)
• Et à des kystes à paroi fine (50 %)
• Nodules et des condensations alvéolaires
plus rares
• Pas d’atteinte médiastinale ganglionnaire
ou d’atteinte pleurale
*PNEUMO 5/2003 20/11/03 11:25 Page 206

pronostic des fibroses pulmonaires idiopathiques et l’effet
potentiel du traitement anti-inflammatoire, les praticiens sont
souvent tentés d’entreprendre une corticothérapie. Cependant,
l’efficacité de ce traitement est limitée et ne peut survenir que
si une fibrose irréversible ne s’est pas développée. De plus, les
effets indésirables ne sont pas négligeables, surtout en présence
de comorbidités. Il existe un consensus (qui ne repose cepen-
dant que sur de très rares essais) pour conseiller un traitement
combinant corticostéroïdes et azathioprine ou cyclophospha-
mide. Après six mois de traitement, soit une aggravation est
constatée et le traitement doit être interrompu ou modifié, soit
il existe une amélioration ou une stabilité et le traitement doit
être poursuivi dans les mêmes conditions. Des thérapeutiques
antifibrosantes ont été récemment proposées ; c’est le cas de
l’interféron gamma qui, dans une étude portant sur 18 patients,
a donné des résultats semblant prometteurs. Ces résultats res-
tent toutefois préliminaires et justifient d’être documentés par
d’autres travaux sur un plus grand nombre de malades.
QUI BÉNÉFICIE DE LA TRANSPLANTATION PULMONAIRE
DANS LES ÉVOLUTIONS FIBROSANTES ?
d’après le résumé de P. Bonette
(chirurgie thoracique et transplantation pulmonaire,
hôpital Foch, Suresnes)
La transplantation pulmonaire est une technique de survie dans
les évolutions terminales de certaines fibroses. Dans le registre
français de l’Établissement français des greffes, la survie à 5 ans
pour les fibroses est de 40 %. Dans le registre international, un
cinquième des transplantations unipulmonaires sont réalisées
pour fibrose, contre seulement 8 % des transplantations bipul-
monaires. Le greffé est menacé par les complications chirurgi-
cales (dysfonction primaire du greffon, complications anasto-
motiques, paralysie phrénique, gastroparésie), les infections
(virales et fongiques surtout), les rejets aigus (surtout la pre-
mière année), la diminution progressive de la fonction respira-
toire sous forme d’une bronchiolite oblitérante, enfin les com-
plications du traitement immunosuppresseur (atteinte rénale,
HTA, ostéoporose, etc.). Le résultat fonctionnel est habituelle-
ment excellent chez les survivants, avec une activité quotidienne
non limitée pour plus de 80 % des patients en vie à long terme.
Les patients qui bénéficient de ces transplantations doivent être
non fumeurs ; ils doivent avoir un âge inférieur à 60 ans, une
corticothérapie réduite à moins de 20 mg/jour lors de l’inscrip-
tion, et une fonction rénale non ou peu altérée ; ils ne doivent
pas avoir d’ostéoporose fracturaire, de coronaropathie instable,
d’insuffisance ventriculaire gauche ou de cancer récent. Ils peu-
vent avoir des antécédents chirurgicaux thoraciques (biopsie
pulmonaire ou symphyse pleurale). Il est souhaitable qu’ils
observent leur traitement, et soient très déterminés à accepter
les contraintes d’une greffe. L’inscription doit être enregistrée
quand la mortalité spontanée de la maladie semble dépasser la
mortalité post-greffe, soit un pronostic vital estimé à moins de
deux ans. Les critères d’indication à une transplantation dans
les fibroses reconnus dans la littérature sont une dyspnée
d’aggravation régulière, avec échec d’un traitement médical
bien conduit, une capacité vitale inférieure à 60-70 % de la
valeur théorique, un rapport TLCO/VA inférieur à 50-60 % de
la valeur théorique, une PO2inférieure à 55 mmHg, une PACO2
supérieure à 45 mmHg, et une HTAP secondaire. À noter que,
dans le cadre des fibroses terminales, la transplantation n’est
pas recommandée dans un contexte de ventilation mécanique,
car la mortalité postopératoire est alors considérablement
accrue. Le bénéfice de la transplantation apparaît important dans
de nombreuses publications.
BIOLOGIE DES MÉTASTASES
d’après le résumé de E. Pujade-Lauraine
(département d’hématologie et d’oncologie médicale,
Hôtel-Dieu, Paris)
La formation de métastases est un processus actif et complexe.
Toutes les tumeurs ne sont pas dotées des fonctions nécessaires
à ce processus. La cellule qui a acquis le pouvoir métastatique
est elle-même le produit d’une évolution clonale de la tumeur.
Les différentes étapes en cause sont l’invasion tumorale, la
migration cellulaire, l’entrée dans les vaisseaux, la greffe des
cellules tumorales dans un organe distant. Ensuite, il y a crois-
sance des cellules métastatiques dans un organe distant grâce à
des substances nutritives fabriquées dans les vaisseaux dirigés
vers la cellule tumorale (processus complexe de néoangioge-
nèse tumorale nécessitant la sécrétion coordonnée de nombreux
facteurs, en tête desquels se situe le VEGF, ou Vascular Endo-
thelial Growth Factor) et grâce à la présence des facteurs né-
cessaires à la stimulation de sa reproduction comme l’EGF
(Epidermal Growth Factor).
Une des tâches essentielles du clinicien au cours du traitement
d’une tumeur maligne localisée est d’essayer de prédire sa capa-
cité métastatique. Les facteurs de dissémination métastatique
sont résumés dans le tableau II. Le résultat de cette apprécia-
tion du risque de métastase infraclinique grâce à tous ces fac-
teurs n’est pas quantitatif, c’est-à-dire qu’il ne permet pas de
préciser si un ou plusieurs organes seront atteints et, si un organe
est atteint, combien de foyers métastatiques infracliniques sont
présents. Néanmoins, on peut penser que plus les facteurs de
risque s’accumulent, plus le risque est grand que les foyers
métastatiques soient nombreux. Cette considération peut avoir
son importance lorsqu’on envisage le traitement chirurgical de
métastases.
LA CHIRURGIE DES MÉTASTASES PULMONAIRES :
PRINCIPALES INDICATIONS ET RÉSULTATS
d’après le résumé de J.F. Regnard
(service de chirurgie thoracique,
Hôtel-Dieu, Paris)
Le registre international des métastases pulmonaires a permis de
colliger 5 206 patients opérés dans un but curatif. Les survies après
résection complète ont été respectivement de 36 et 26 % à 5 et
10 ans. La médiane de survie a été de 35 mois. Les meilleurs résul-
tats ont été observés dans les tumeurs germinales (survie à 10 ans
de 63 %) et les plus défavorables dans les mélanomes (survie à
207
La Lettre du Pneumologue - Volume VI - no5 - septembre-octobre 2003
*PNEUMO 5/2003 20/11/03 11:25 Page 207
 6
7
8
6
7
8
1
/
8
100%