Aldostérone, spironolactone et insuffisance cardiaque P

136
La Lettre du Pharmacologue - Volume 15 - n
os
7-8 - septembre-octobre 2001
PHARMACOLOGIE
algré l’amélioration de la prise en charge de l’in-
suffisance cardiaque, la morbidité et la morta-
lité associées à cette pathologie demeurent éle-
vées (1, 2). De plus, les perspectives d’évolution suggèrent que
la prévalence de l’insuffisance cardiaque devrait augmenter de
façon importante au cours des années à venir, du fait du vieillis-
sement de la population et de l’amélioration des thérapeutiques,
notamment dans le domaine de l’ischémie myocardique et de
l’hypertension artérielle (3).
Les progrès les plus marquants dans le domaine du traitement
médicamenteux de l’insuffisance cardiaque ont tous porté sur
les interactions avec les grands systèmes neuro-hormonaux :
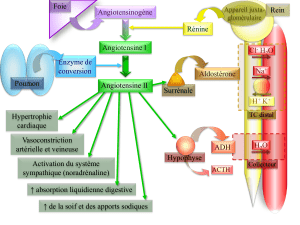
système sympathique, endothéline et surtout système rénine-
angiotensine-aldostérone (SRAA). S’il a longtemps été consi-
déré que la plupart des effets des bloqueurs du SRAA résul-
taient de l’inhibition des effets de l’angiotensine II, il est clair
aujourd’hui que le blocage des effets de l’aldostérone joue éga-
lement un rôle important, ce que montrent aussi indirectement
les observations démontrant le rôle délétère certain de l’aldo-
stérone dans l’insuffisance cardiaque.
Dans ce contexte, il a été montré que le traitement chronique
par les inhibiteurs de l’enzyme de conversion de l’angioten-
sine I (IEC) ou les antagonistes des récepteurs AT1de l’angio-
tensine II conduit à une inhibition incomplète de la production
d’aldostérone. Les conséquences d’un tel phénomène d’échap-
pement et la connaissance des effets délétères de l’aldostérone
justifient l’utilisation d’un antagoniste des récepteurs de
l’aldostérone, et en particulier de la spironolactone, dans le
traitement de l’insuffisance cardiaque.
Cet article résume nos connaissances actuelles dans le domaine
des effets cardiovasculaires de l’aldostérone, son rôle dans l’in-
suffisance cardiaque et les résultats obtenus avec la spirono-
lactone dans cette pathologie.
Aldostérone, spironolactone et insuffisance cardiaque
!
V. Richard*, C. Thuillez*
*Service de pharmacologie, INSERM E9920, CHU de Rouen, 76031 Rouen
Cedex.
RÉSUMÉ.
L’aldostérone est le principal minéralocorticoïde de l’organisme. Elle est produite par les cellules glomérulées de la corticosur-
rénale, mais également au niveau cardiaque (au moins dans les modèles expérimentaux). Les principaux effets biologiques de l’aldostérone
s’expriment au niveau rénal (stimulation de la réabsorption de sodium et d’eau et de l’excrétion de potassium et de magnésium), mais égale-
ment au niveau neuronal (inhibition du recaptage des catécholamines) et au niveau cardiaque (en particulier développement de fibrose). Ces
effets dépendent de la stimulation d’un récepteur nucléaire, mais aussi probablement d’un récepteur membranaire, responsable des effets
immédiats de l’aldostérone.
La synthèse d’aldostérone est augmentée de façon importante dans l’insuffisance cardiaque, en particulier suite à l’activation du système rénine-
angiotensine. Si l’on se réfère aux effets de l’aldostérone au niveau rénal et cardiaque, il est clair que le blocage de la production et/ou des effets
de l’aldostérone pourrait exercer des effets bénéfiques dans l’insuffisance cardiaque, en particulier en termes d’amélioration de fonction
cardiaque et de diminution de l’incidence des troubles du rythme. Néanmoins, de nombreux essais cliniques ont démontré que la production
d’aldostérone était contrôlée de façon très imparfaite par les inhibiteurs de l’enzyme de conversion ou les antagonistes des récepteurs de l’an-
giotensine II. L’existence d’un tel échappement de l’aldostérone dans l’insuffisance cardiaque ainsi que le développement des connaissances dans
la physiopathologie de ce facteur ont conduit à l’évaluation des effets d’un antagoniste des récepteurs de l’aldostérone, la spironolactone, dans
l’insuffisance cardiaque. Ainsi, l’étude multicentrique RALES (Randomized Aldactone Evaluation Study) a clairement démontré le bénéfice de la
spironolactone chez des patients en insuffisance cardiaque sévère, en particulier en termes de sévérité de l’insuffisance cardiaque et de
mortalité. Le blocage des récepteurs de l’aldostérone paraît donc constituer un apport majeur dans le traitement de l’insuffisance cardiaque, au
moins dans un contexte de dysfonction ventriculaire sévère et en l’absence d’insuffisance rénale et d’hyperkaliémie.
Mots-clés :
Rénine - Angiotensine - Aldostérone -Insuffisance cardiaque - Spironolactone.
M

La Lettre du Pharmacologue - Volume 15 - n
os
7-8 - septembre-octobre 2001
137
PHARMACOLOGIE
SYNTHÈSE ET MÉTABOLISME DE L’ALDOSTÉRONE
Le principal site de synthèse d’aldostérone est la glande corti-
cosurrénale, et, en particulier, les cellules glomérulées situées
immédiatement sous la capsule. Les cellules surrénaliennes ne
stockent pas l’aldostérone ; au contraire, elles sont capables de
la synthétiser extrêmement rapidement au niveau des mito-
chondries.
La synthèse d’aldostérone se fait à partir de cholestérol
(figure 1). Celui-ci est mobilisé à partir des réserves consti-
tuées par des gouttelettes lipidiques, et transformé en prégné-
nolone par la cholestérol desmolase, ce qui constitue une étape
précoce de régulation de la synthèse. La prégnénolone est
ensuite convertie en progestérone, elle-même hydroxylée en
11-désoxycorticostérone (DOC), puis la DOC est hydroxylée
en corticostérone par la cytochrome P-450 11β/18β-hydroxy-
lase. La conversion corticostérone-aldostérone se fait alors
grâce à la cytochrome P-450 aldostérone synthétase. Cette
conversion met d’abord en jeu une hydroxylation (conduisant
à la production de 18-OH corticostérone), suivie de la forma-
tion d’un aldéhyde qui constitue l’aldostérone, ces deux étapes
mettant en jeu l’aldostérone synthétase.
Au contraire des glucocorticoïdes, il n’existe pas de protéine
de transport spécifique de l’aldostérone. Plus de 50 % de l’al-
dostérone circulante est non liée, et le volume de distribution
de cette hormone est très important (35 litres pour une personne
de 70 kg).
Le principal site de métabolisme de l’aldostérone est le foie,
qui en inactive 85 % en un seul passage. La tétrahydroaldosté-
rone (glucuronoconjuguée) constitue le principal métabolite
urinaire. La vitesse de dégradation de l’aldostérone dépend du
débit hépatique et de son extraction au niveau des cellules paren-
chymateuses, deux paramètres qui peuvent être modifiés dans
l’insuffisance cardiaque. Ainsi, les modifications de débit hépa-
tique et de métabolisme associées à l’insuffisance cardiaque
peuvent conduire à une augmentation par un facteur 3 des
concentrations sanguines et de la demi-vie de l’aldostérone.
L’hyperaldostéronisme rencontré dans l’insuffisance cardiaque
est donc lié à la fois à l’activation du SRAA et à la diminution
de la clairance hépatique (4).
De nombreux facteurs peuvent réguler la synthèse d’aldosté-
rone au niveau surrénalien. Les principaux stimulants de la syn-
thèse d’aldostérone sont :
"L’angiotensine II, qui en constitue le stimulant majeur. Au
contraire de l’ACTH, la stimulation induite par l’angiotensine II
persiste en cas de stimulation prolongée. Ainsi, certains effets
de l’angiotensine II (par exemple, la stimulation de la
réabsorption de Na+) s’exercent à la fois de façon directe et par
l’intermédiaire de la sécrétion d’aldostérone. L’angiotensine II
stimule la sécrétion d’aldostérone par l’intermédiaire de la
stimulation des récepteurs AT1,conduisant à une activation
de la phospholipase C et à une augmentation de calcium intra-
cellulaire qui stimule les enzymes de biosynthèse calcium-
dépendantes (5).
"L’ACTH, dont les effets sur la stimulation d’aldostérone sont
temporaires (de l’ordre de 24 heures), avec un retour à la nor-
male qui peut s’observer même en cas de taux élevés d’ACTH.
Cette hormone joue un rôle important sur le cycle nycthémé-
ral de sécrétion d’aldostérone.
"La kaliémie, qui constitue un système de régulation sensible
de la sécrétion d’aldostérone. Cette régulation par le K+est par
ailleurs influencée par la balance sodée, une balance sodée
négative augmentant la sensibilité au K+.
Au-delà de cette description “classique” des stimulants de la
sécrétion d’aldostérone, d’autres facteurs ont été potentielle-
ment impliqués : c’est notamment le cas de l’endothéline. En
effet, on trouve des récepteurs de l’endothéline au niveau des
cellules glomérulées de la corticosurrénale. Ces récepteurs sont
de type ETBchez le rat et ETAet ETBchez l’homme. La
stimulation de ces récepteurs provoque non seulement une
augmentation de la production d’aldostérone, mais aussi une
stimulation de la prolifération des cellules glomérulées (6).
À l’inverse, la synthèse d’aldostérone peut être inhibée par le
facteur atrial natriurétique (par un effet direct qui s’ajoute à
l’effet indirect lié à l’inhibition de la synthèse de rénine), l’hé-
parine, la ouabaïne, la dopamine et les inhibiteurs de la stéroï-
dogenèse (dont les androgènes).
Récemment, de nombreuses données ont suggéré l’existence
d’une synthèse locale d’aldostérone au niveau extrarénal. C’est,
en particulier, le cas du cerveau (7), des vaisseaux (8, 9), mais
aussi du myocyte cardiaque, au moins chez le rat (10-12). Au
niveau cardiaque, bien que les facteurs régulant la synthèse
d’aldostérone restent à déterminer de façon précise, il est
probable que l’angiotensine II (notamment celle produite au
niveau tissulaire) joue là aussi un rôle majeur (12).
AII
ACTH
K+
ANF
Cholestérol
Prégnénolone
Progestérone
Désoxycorticostérone (DOC)
Aldostérone
AT1ETA-ETB
ET-1
Aldostérone
synthétase
Corticostérone
Na+
K+
Mitochondrie
(Cellule glomérulée, cœur, artère)
10
+
+
+
-+10
Figure 1.Voies de synthèse de l’aldostérone.

138
La Lettre du Pharmacologue - Volume 15 - n
os
7-8 - septembre-octobre 2001
PHARMACOLOGIE
EFFETS BIOLOGIQUES DE L’ALDOSTÉRONE
L’aldostérone est le minéralocorticoïde le plus puissant de
l’organisme. Comme son nom l’indique, ce stéroïde intervient
dans la régulation des minéraux, et tout particulièrement dans
la réabsorption du Na+et de l’eau (aux dépens de l’excrétion
de K+et Mg2+). Cependant, outre ces effets “classiques”,
l’aldostérone peut intervenir au niveau de tissus non épithé-
liaux, essentiellement au niveau cardiaque.
Les principaux effets biologiques de l’aldostérone et leurs
conséquences au niveau du système cardiovasculaire sont résu-
més dans le tableau I.
Effets rénaux
Les effets majeurs de l’aldostérone se situent au niveau du
tubule distal du néphron. À ce niveau, le potassium filtré a été
entièrement réabsorbé, tandis que le sodium l’a été à 80-90 %.
Le tube distal est le site majeur de la régulation finale de
l’excrétion de sodium et de potassium, en particulier par des
échanges sodium contre potassium ou proton. L’aldostérone
stimule la réabsorption de sodium et l’excrétion de potassium.
La réabsorption de sodium crée un gradient électrochimique
conduisant à un transfert passif d’ions chlore, ainsi qu’une
sécrétion d’ions H+.
Les effets cellulaires de l’aldostérone dépendent de sa liaison
avec un récepteur intracellulaire spécifique, le récepteur miné-
ralocorticoïde. Le couplage hormone/récepteur va induire une
migration vers le noyau et une fixation sur des séquences spé-
cifiques de l’ADN. Il s’ensuit une augmentation de la synthèse
des protéines responsables de l’échange Na+/K+. Cette stimu-
lation se fait au niveau transcriptionnel, et l’inhibition de la syn-
thèse protéique abolit les effets de l’aldostérone. Parmi les pro-
téines dont la synthèse est augmentée par l’aldostérone, on peut
citer la Na+/K+ATPase,le canal Na+sensible à l’amiloride, ainsi
que des enzymes impliquées dans le métabolisme énergétique,
et probablement d’autres échangeurs ioniques. Ces mécanismes
d’action cellulaire de l’aldostérone, qui nécessitent une
synthèse protéique, expliquent le délai observé dans les effets
de l’aldostérone.
Bien que le récepteur nucléaire ne soit pas sélectif vis-à-vis de
l’aldostérone et soit également capable de fixer les glucocorti-
coïdes, la sélectivité vis-à-vis de l’aldostérone est assurée par
la présence de 11βhydroxystéroïde déshydrogénase, qui
dégrade les glucocorticoïdes en dérivés présentant une affinité
faible pour le récepteur (13).
Effets cardiovasculaires
La présence de récepteurs minéralocorticoïdes dans le myocarde
et sur les vaisseaux est maintenant bien établie. On retrouve ce
récepteur au niveau des myocytes humains (oreillette et ventricule)
(14) ainsi que sur les cellules endothéliales et musculaires lisses
de gros troncs artériels (aorte, artère pulmonaire, carotide, mésen-
térique) (15). En revanche, ces récepteurs n’ont pas été détectés
dans les artérioles, les capillaires et les veines (15). Le myocyte
cardiaque possède également une activité 11βhydroxystéroïde
déshydrogénase, qui est responsable de la sélectivité du récepteur
nucléaire vis-à-vis de l’aldostérone. Ainsi, la présence à la fois des
enzymes de synthèse et de dégradation et des récepteurs de l’al-
dostérone plaide pour l’existence d’un système aldostérone local
dans le muscle cardiaque et les cellules vasculaires, comme cela
a été démontré avec le système rénine-angiotensine.
Dans le système cardiovasculaire, la stimulation des échanges
ioniques par l’aldostérone est associée à une modification des
concentrations ioniques de la cellule ou de son environnement extra-
cellulaire immédiat. Cependant, les cibles cellulaires exactes de l’al-
dostérone au niveau des tissus non épithéliaux restent à déterminer.
Indépendamment des effets ioniques, de nombreuses données
suggèrent que l’aldostérone exerce au niveau cardiaque et vas-
culaire un puissant effet stimulant de la synthèse de collagène
et de la fibrose. Ainsi, chez le rat, la perfusion d’aldostérone à
des doses n’induisant pas d’hypertension artérielle conduit au
développement d’une fibrose interstitielle et périvasculaire (16).
En outre, l’aldostérone stimule la synthèse de collagène sur des
fibroblastes en culture (17).Ces observations plaident en faveur
d’un effet direct de l’aldostérone, indépendant du contexte
hémodynamique et, au moins en partie, de l’activation du sys-
tème rénine-angiotensine. La stimulation de la fibrose dans les
situations où les concentrations d’aldostérone sont augmentées
peut avoir des conséquences majeures, à la fois sur le cœur (en
termes de modification de compliance et d’incidence de
troubles du rythme) et les vaisseaux (diminution de compliance
et augmentation de pression artérielle) (tableau II).
#$Na
+
"hypertrophie ventriculaire gauche
"%compliance artérielle
#%K
+
"troubles du rythme
#%Mg
2+
"troubles du rythme
#Potentialisation du tonus sympathique
"effet proarythmique
#Fibrose "cardiaque – compliance ventriculaire gauche
– troubles du rythme
"vasculaire – mécanique artérielle
(compliance, pression artérielle systolique)
#$volémie "pression artérielle
"volume et mécanique cardiaques
"stress pariétal
Tableau I. Effets biologiques de l’aldostérone et conséquences
cardiovasculaires.
&Conséquences liées à la rigidité du collagène
– diminution de l’élasticité musculaire
– augmentation de la rigidité diastolique
– gêne au remplissage passif
– augmentation du remplissage actif télédiastolique
'Conséquences dues à une hétérogénéité tissulaire
– dysfonctionnement systolique
– création de circuits de réentrée
– augmentation de l’automaticité
– hétérogénéité de la propagation
(Conséquences dues à la perte de substance contractile
– diminution de la tension active
Tableau II. Principales conséquences de la fibrose ventriculaire.

La Lettre du Pharmacologue - Volume 15 - n
os
7-8 - septembre-octobre 2001
139
PHARMACOLOGIE
Les effets vasculaires de l’aldostérone pourraient enfin
dépendre de la stimulation d’un récepteur membranaire de
l’aldostérone, distinct du récepteur nucléaire (18). Les effets de
l’activation de ces récepteurs seraient rapides, ne nécessitant
pas de synthèse protéique. Les mécanismes de transduction
impliqués lors de la stimulation de ces récepteurs membranaires
impliquent la stimulation de l’échangeur sodium/protons, mais
aussi l’activation de la voie de la phospholipase C, se tradui-
sant en particulier par un effet vasoconstricteur (19).
ALDOSTÉRONE ET INSUFFISANCE CARDIAQUE
Insuffisance cardiaque et concentrations circulantes d’aldo-
stérone
L’activation de différents systèmes neurohumoraux constitue
un élément déterminant du développement de l’insuffisance
cardiaque chronique. Parmi les systèmes activés, on note le
système sympathique, la vasopressine, le facteur atrial natriu-
rétique, le système endothéline et, bien entendu, le système
rénine-angiotensine-aldostérone.
L’hyperaldostéronisme observé dans l’insuffisance cardiaque
est de nature secondaire, et résulte principalement de l’activa-
tion de la production de rénine, bien qu’il soit probable que
d’autres facteurs humoraux y participent. Ce peut être le cas de
l’endothéline, bien que les relations entre endothéline et aldo-
stérone dans l’insuffisance cardiaque restent à étudier précisé-
ment. Par ailleurs, certains traitements de l’insuffisance car-
diaque peuvent affecter la synthèse d’aldostérone. C’est le cas
notamment des diurétiques, principalement par l’augmentation
de libération de rénine qu’ils induisent.
Indépendamment des modifications de synthèse, l’augmenta-
tion des taux d’aldostérone dans l’insuffisance cardiaque pour-
rait être liée en partie à une moindre élimination du minéralo-
corticoïde. En effet, comme cela a déjà été mentionné,
l’élimination de l’aldostérone est principalement hépatique, et
donc proportionnelle au débit sanguin hépatique. Ainsi, chez
des patients insuffisants cardiaques, il a été démontré que la
baisse de débit hépatique provoque une augmentation de la
demi-vie d’élimination de l’aldostérone (20).
La présence de nombreux facteurs confondants, tels que la
charge sodée et les traitements associés, rend difficile l’éva-
luation exacte de l’augmentation des taux d’aldostérone chez
des patients insuffisants cardiaques. Cependant, plusieurs
études anciennes ont pu évaluer ces taux chez des patients non
traités. Par exemple, dans une analyse d’un sous-groupe de
l’étude CONSENSUS II, une augmentation des taux d’aldo-
stérone a été notée après un infarctus, uniquement chez les
patients présentant une insuffisance cardiaque (21). Une aug-
mentation a également été mesurée chez des patients en insuf-
fisance cardiaque sévère (classe III et IV de la NYHA) (22).
Les relations entre concentrations d’aldostérone et morbi-mor-
talité dans l’insuffisance cardiaque ont aussi été étudiées dans
l’étude CONSENSUS I, chez 239 patients en stade IV d’in-
suffisance cardiaque et randomisés dans le bras placebo. Tous
ces patients étaient traités par un diurétique, 94 % recevaient
des digitaliques et la moitié un vasodilatateur (en général le
dinitrate d’isosorbide). Les résultats de cette étude démontrent
une corrélation significative entre les concentrations d’aldo-
stérone et la mortalité évaluée à 6 mois (p = 0,003) (23).
Insuffisance cardiaque et production cardiaque d’aldostérone
Des données expérimentales récentes suggèrent que l’insuffi-
sance cardiaque affecte la production cardiaque d’aldostérone.
En effet, dans un modèle d’infarctus du myocarde chez le rat,
on observe une augmentation marquée de l’expression des
gènes codant pour l’aldostérone synthétase, associée à une aug-
mentation importante de la production d’aldostérone au niveau
du myocarde non nécrosé, et ce sans modification des taux plas-
matiques d’aldostérone ni de l’expression d’aldostérone syn-
thétase au niveau surrénalien (12). Cette activation s’accom-
pagne d’une augmentation de production d’angiotensine II, et
est abolie par un antagoniste des récepteurs AT1de l’angioten-
sine II. Cela suggère que dans une situation d’infarctus de myo-
carde associé à une dysfonction ventriculaire gauche modérée,
il existe une activation locale de la synthèse cardiaque d’aldo-
stérone, et ce même en l’absence de stimulation de la produc-
tion surrénalienne. L’existence d’une telle activation tissulaire
reste toutefois à démontrer de façon directe chez l’homme.
Conséquences de l’augmentation d’aldostérone dans l’insuffi-
sance cardiaque
L’augmentation des concentrations circulantes d’aldostérone
facilite la rétention hydrosodée et participe donc au dévelop-
pement d’œdème et de congestion observables en pratique cli-
nique. Chez les patients en insuffisance cardiaque, la surcharge
volumique tend à aggraver le contexte hémodynamique. La
baisse de débit cardiaque qui en résulte s’accompagne d’une
diminution du débit sanguin rénal et d’une activation supplé-
mentaire du système rénine-angiotensine-aldostérone, contri-
buant au développement d’un véritable cercle vicieux.
En parallèle de la rétention hydrosodée, l’augmentation de
sécrétion de Mg2+ et de K+induite par l’aldostérone aggrave la
déplétion en magnésium et l’hypokaliémie, et particulièrement
celles induites par les diurétiques. Les conséquences princi-
pales de ces modifications ioniques se détectent au niveau car-
diaque, où elles participent à l’instabilité électrique et tendent
donc à augmenter les arythmies (24, 25.)
Au niveau cardiaque, les conséquences de l’hyperaldostéro-
nisme se traduisent principalement en termes de stimulation de
développement d’hypertrophie et de fibrose. En effet, comme
il a déjà été indiqué, la stimulation des récepteurs minéralo-
corticoïdes présents sur les fibroblastes cardiaques conduit à
une augmentation de la synthèse de collagène par ces cellules
(16, 17). La fibrose myocardique, qu’elle soit interstitielle ou
périvasculaire, peut perturber la perfusion myocardique et dimi-
nuer la compliance ventriculaire gauche, contribuant à la
dysfonction contractile (26) (tableau II). Par ailleurs, l’hété-
rogénéité de conduction associée au développement de zones

140
La Lettre du Pharmacologue - Volume 15 - n
os
7-8 - septembre-octobre 2001
PHARMACOLOGIE
focales de fibrose peut augmenter l’incidence des troubles du
rythme ventriculaire. Cet effet proarythmique peut être renforcé
par l’inhibition par l’aldostérone du recaptage myocardique des
catécholamines (27). Enfin, l’aldostérone étant capable d’alté-
rer les réponses baroréflexes (au moins chez le volontaire sain)
(28), il est possible que l’augmentation d’aldostérone dans
l’insuffisance cardiaque joue un rôle dans la désensibilisation
du baroréflexe caractéristique de cette pathologie.
Au niveau vasculaire, les effets vasoconstricteurs de l’aldosté-
rone, qu’ils soient directs ou secondaires à la stimulation sym-
pathique, peuvent contribuer à augmenter les résistances péri-
phériques, et donc à aggraver le contexte d’insuffisance
cardiaque. De plus, le développement de la fibrose des gros
troncs pourrait contribuer à diminuer leur compliance (29).
Enfin, les effets vasculaires de l’aldostérone impliquent égale-
ment la circulation veineuse. Ainsi, une étude réalisée chez
16 insuffisants cardiaques traités par captopril, digoxine et furo-
sémide démontre une relation inverse entre aldostérone et capa-
citance veineuse, suggérant un rôle direct de l’aldostérone sur
la précharge (30).
SPIRONOLACTONE ET INSUFFISANCE CARDIAQUE
Compte tenu du fait que le principal stimulant de la sécrétion
d’aldostérone est l’angiotensine II, il paraît normal de penser
que les inhibiteurs de l’enzyme de conversion de l’angioten-
sine I, ou encore les antagonistes des récepteurs AT1de l’an-
giotensine II, diminuent la production d’aldostérone dans l’in-
suffisance cardiaque, cet effet inhibiteur ayant été retrouvé dans
les grands essais cliniques. Par exemple, dans l’étude
CONSENSUS, un traitement de 6 mois par l’énalapril conduit
à une réduction de plus de 50 % des concentrations plasma-
tiques d’aldostérone (23). Cependant, les études réalisées avec
les inhibiteurs de l’enzyme de conversion chez l’insuffisant
cardiaque s’accordent à dire que l’inhibition de la production
d’aldostérone en réponse à ces traitements est incomplète (31).
Dans de nombreux cas, on observe même un retour aux valeurs
de base des concentrations d’aldostérone en présence d’un
traitement chronique par un IEC (32).
Les mécanismes de phénomène “d’échappement” de l’al-
dostérone pourraient être liés en partie au blocage incom-
plet de la production d’angiotensine II par les IEC en trai-
tement chronique. Toutefois, des résultats récents
démontrent que le phénomène d’échappement de l’aldosté-
rone se retrouve avec les antagonistes des récepteurs AT1.
Par exemple, dans l’étude pilote RESOLVD (33), aucune
modification des taux d’aldostérone n’a été observée après
43 semaines de traitement par un IEC (l’énalapril), un anta-
goniste des récepteurs AT1(le candesartan), ou leur combi-
naison. D’autres mécanismes pourraient donc expliquer cet
échappement de l’aldostérone, en particulier le fait que,
comme il a été indiqué plus haut, la sécrétion d’aldostérone
est sous la dépendance de nombreux facteurs, indépendam-
ment de l’angiotensine II (ACTH, potassium, endothéline,
ANF, débit hépatique, etc.).
Compte tenu des effets délétères de l’aldostérone et du contrôle
incomplet de sa sécrétion par les IEC ou les antagonistes AT1,
l’utilisation de médicaments bloquant directement la produc-
tion ou les effets tissulaires de l’aldostérone paraissait promet-
teuse. La principale substance utilisée dans ce contexte est la
spironolactone, un antagoniste compétitif du récepteur miné-
ralocorticoïde de l’aldostérone.
Paradoxalement, peu d’études ont évalué les effets de la spiro-
nolactone dans des modèles expérimentaux d’insuffisance car-
diaque. Cependant, il a récemment été montré que cet antago-
niste était capable d’inhiber la fibrose myocardique dans un
modèle d’infarctus du myocarde chez le rat, et ce sans modifi-
cation de pression artérielle, bien que, dans ce modèle, l’effet de
la spironolactone soit inférieur à celui d’un antagoniste AT1(12).
L’efficacité de la spironolactone chez les patients insuffisants
cardiaques a d’abord été évaluée dans des études cliniques de
petite taille et sur des périodes courtes. Tsutamoto et al. (34)
ont ainsi montré que la spironolactone diminuait l’extraction
transcardiaque d’aldostérone, elle-même corrélée à un mar-
queur biologique de fibrose cardiaque. Au plan hémodyna-
mique, une étude randomisée en double aveugle réalisée chez
31 insuffisants cardiaques traités par un IEC et un diurétique
(35) a démontré que la spironolactone (50-100 mg/j) induisait
une baisse de fréquence cardiaque, ainsi qu’une augmentation
de la variabilité de fréquence cardiaque, reconnue comme étant
un facteur pronostique dans l’insuffisance cardiaque. En paral-
lèle, cette étude rapportait également une diminution des taux
sériques d’un marqueur du collagène cardiaque.
Les effets de la spironolactone sur l’équilibre électrolytique et
les modifications électrocardiographiques ont été évalués chez
42 patients insuffisants cardiaques en classe II et III de la
NYHA et traités par IEC + diurétique (27). On observe lors du
traitement par la spironolactone une diminution du nombre
d’extrasystoles ventriculaires évalué par holter ; cette diminu-
tion est inversement corrélée à l’augmentation du magnésium
plasmatique. Ces propriétés antiarythmiques de la spironolac-
tone ont aussi été retrouvées dans une seconde étude randomisée
réalisée chez 35 patients en classe III de la NYHA (36). On
retrouve dans cette étude une diminution du nombre d’extra-
systoles ventriculaires et des épisodes de tachycardie.
L’efficacité de la spironolactone a également été vérifiée chez
16 patients (classe III de la NYHA) non répondeurs au traite-
ment IEC-diurétique de l’anse. Chez ces patients, on note une
augmentation importante de la natriurèse et de la diurèse, asso-
ciée à une diminution de poids, et une amélioration des symp-
tômes pour 81 % d’entre eux (37).
Les effets de la spironolactone dans l’insuffisance cardiaque ne
se limitent pas au cœur et au rein, mais s’étendent aussi au sys-
tème vasculaire. Ainsi, une étude récente suggère qu’un traite-
ment d’un mois par la spironolactone chez 10 patients insuffi-
sants cardiaques améliore la fonction endothéliale périphérique,
évaluée par la réponse vasodilatatrice à l’acétylcholine et la
réponse vasoconstrictrice à un inhibiteur de synthèse de NO
 6
7
6
7
1
/
7
100%