Spironolactone et insuffisance cardiaque chronique

ACTUALITÉS
La Lettre du Cardiologue - n° 322 - décembre 1999
6
Résumé
✔
L’évolution de l’insuffisance cardiaque chronique reste grave, ce qui justifie la poursuite de la recherche pour améliorer les
stratégies de traitement. Ce traitement est actuellement inspiré par la théorie neurohormonale. Celle-ci rend responsables de
l’aggravation clinique les cercles vicieux engendrés par la stimulation excessive des systèmes angiotensine-aldostérone et
adrénergique.
✔
Les inhibiteurs de l’enzyme de conversion de l’angiotensine ont démontré leur efficacité en réduisant l’activité du système
angiotensine-aldostérone, et en diminuant la mortalité. Cependant, cette dernière reste élevée, en partie expliquée par l’échap-
pement de la sécrétion d’aldostérone lors du traitement par les inhibiteurs de l’enzyme de conversion. L’aldostérone a des
effets délétères, en provoquant la baisse de la kaliémie et de la magnésémie, favorisant ainsi les arythmies graves. Elle contri-
bue à la rétention hydrosodée et à l’hypervolémie, qui aggrave la fonction ventriculaire gauche.
✔
Un inhibiteur spécifique de l’aldostérone, la spironolactone, peut combattre ces effets. Toutefois, son association au traite-
ment incontournable que sont devenus les inhibiteurs de l’enzyme de conversion expose au risque d’induire une hyperkalié-
mie. Des doses faibles de spironolactone, voisines de 25 mg par jour, sont généralement bien supportées, mais justifient une
surveillance régulière de la kaliémie. Des doses plus fortes peuvent être transitoirement utilisées en cas d’insuffisance car-
diaque rebelle ou sévère, à condition de surveiller encore plus fréquemment la kaliémie.
✔
L’étude RALES, destinée à rechercher une amélioration clinique grâce à la spironolactone ajoutée au traitement habituel de
l’insuffisance cardiaque chronique, a été arrêtée en raison d’un effet favorable significatif sur la survie. Cependant, d’autres
études seront nécessaires pour préciser la place exacte de la spironolactone dans le blocage des systèmes neurohormonaux,
blocage qui constitue la base du traitement moderne de l’insuffisance cardiaque.
Mots-clés : Spironolactone - IEC - Insuffisance cardiaque.
insuffisance cardiaque est une affection fréquente chez
les personnes âgées, et sa prévalence dans la popula-
tion est croissante en raison de l’augmentation de la
durée de la vie. Ses causes principales sont les cardiopathies isché-
miques, les myocardiopathies dilatées et l’hypertension artérielle,
alors que les cardiopathies rhumatismales sont en déclin (1). Son
pronostic reste grave, et la mortalité à 5 ans, évaluée avant l’ère
du traitement par les inhibiteurs de l’enzyme de conversion (IEC),
atteignait 75 % chez homme et 62 % chez la femme (1). L’avè-
nement des IEC a permis une amélioration significative de son
évolution, sans toutefois suffire à éviter de nombreuses évolu-
tions défavorables (2, 3). Des mécanismes compensateurs com-
plexes entrent en jeu quand la fonction cardiaque systolique et/ou
diastolique est altérée. Certains de ces mécanismes sont néfastes
au long cours, et entraînent une aggravation secondaire de la fonc-
tion cardiaque et de l’évolution clinique. Les recherches phar-
macologiques et cliniques tentent de découvrir de nouveaux trai-
tements, mais aussi de réévaluer des molécules connues à la
lumière de nouvelles conceptions physiopathologiques.
Quelle que soit la cause
initiale de l’altération
de la contractilité myo-
cardique et/ou de la compliance ventriculaire (ischémique, pri-
mitive, hypertensive ou valvulaire), le mécanisme de l’insuffi-
sance cardiaque qui en résulte finalement a connu des explications
Spironolactone et insuffisance cardiaque chronique
●C. Le Pailleur*
*Praticien hospitalier, clinique cardiologique (Pr A. Vacheron), hôpital
Necker, Paris.
L
‘
Physiopathologie
de l’insuffisance cardiaque
chronique
.../...


La Lettre du Cardiologue - n° 322 - décembre 1999
8
différentes selon les époques. Les méthodes thérapeutiques qui
en ont découlé ont, elles aussi, beaucoup varié.
L’ancienne explication cardiorénale, qui privilégiait l’insuffisance
d’élimination hydrosodée par le rein, a fait place dans les années
60 à 80 à la conception hémodynamique, qui accuse l’excès de
vasoconstriction artérielle et veineuse. L’utilisation de vasodila-
tateurs, artériels, veineux ou mixtes en a résulté. Parmi ceux-ci,
les inhibiteurs de l’enzyme de conversion, qui réduisent la sécré-
tion de l’angiotensine II, ont vite démontré leur supériorité sur
tous les autres vasodilatateurs. De multiples études prospectives
et randomisées ont démontré qu’ils améliorent les signes fonc-
tionnels, la fonction cardiaque et la survie (2, 3). On a ainsi
constaté que les vasodilatateurs ne sont pas tous équivalents, et
que leur efficacité n’est pas parallèle à leur pouvoir vasodilata-
teur. D’autre part, la conception hémodynamique a été de nou-
veau mise en échec en raison de l’effet défavorable de plusieurs
produits inotropes positifs au cours de l’insuffisance cardiaque
chronique. Seuls les digitaliques ont prouvé leur intérêt dans ce
cas, alors que beaucoup d’autres produits inotropes positifs ont
eu un effet défavorable. Ainsi, la stimulation de la contractilité
myocardique déprimée, bien que logique selon la conception
hémodynamique, ne donnait pas les résultats espérés.
Toutes ces anomalies, mal expliquées par le modèle hémodyna-
mique, ont conduit à envisager un nouveau modèle physiopatho-
logique de l’insuffisance cardiaque, reposant sur la théorie neu-
rohormonale (4). Selon cette conception, la stimulation des
systèmes rénine-angiotensine-aldostérone et sympathique est res-
ponsable de la vasoconstriction systémique et favorise la réten-
tion hydrosodée. Le traitement doit donc agir directement sur ces
axes neurohormonaux pour avoir une action efficace et persis-
tante. L’insuffisance cardiaque chronique ayant des mécanismes
complexes, son traitement ne peut qu’être multifactoriel, com-
battant les stimulations neurohormonales néfastes. Les succès
confirmés des IEC et, plus récemment, les espoirs que soulève
l’emploi prudent de certains bêtabloquants viennent étayer ces
concepts.
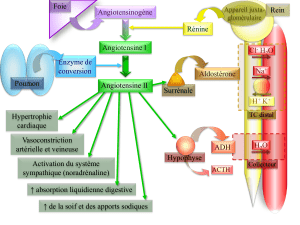
L’aldostérone est sécré-
tée par le cortex des sur-
rénales. Son action prin-
cipale consiste en une
rétention hydrosodée. Au cours de l’insuffisance cardiaque, son
excès de sécrétion contribue à la surcharge hémodynamique du
cœur, aggravant d’autant les signes d’insuffisance cardiaque. De
plus, elle induit une fuite de potassium et de magnésium, ce qui
favorise la survenue de troubles du rythme dangereux et augmente
d’autant le risque de mort subite. D’autre part, elle potentialise
les effets de la noradrénaline. Ces effets théoriques défavorables
se traduisent en clinique par un excès de mortalité quand l’aldo-
stérone plasmatique est élevée, ce qu’illustre la corrélation très
significative entre celle-ci et la mortalité des insuffisants car-
diaques chroniques (5).
Des arguments expérimentaux et cliniques font suspecter d’autres
effets négatifs de l’aldostérone, en particulier celui de favoriser la
fibrose myocardique et d’altérer la régulation du rythme cardiaque
par le système nerveux autonome (6). Des constatations cliniques ont
montré que l’hypertrophie myocardique pathologique est corrélée
très significativement au taux d’aldostérone plasmatique (7). De tels
effets contribuent également à favoriser les arythmies cardiaques.
La sécrétion de l’aldostérone est stimulée par l’angiotensine II.
Cette dernière, qui est un puissant vasoconstricteur, est l’abou-
tissement d’une chaîne qui va de l’angiotensinogène hépatique à
l’angiotensine I sous l’effet de la rénine d’origine rénale, puis de
l’angiotensine I à l’angiotensine II grâce à l’enzyme de conver-
sion de l’angiotensine. Cette dépendance explique pourquoi un
IEC, en réduisant la synthèse de l’angiotensine II, diminue aussi
la sécrétion de l’aldostérone. Ainsi, un traitement par énalapril
diminue en six semaines la sécrétion de presque toutes les hor-
mones impliquées dans le mauvais pronostic de l’insuffisance
cardiaque, en particulier le taux d’aldostérone (5). On pouvait
donc espérer traiter les effets néfastes de l’aldostérone par un IEC,
qui a d’autre part une action favorable sur l’insuffisance cardiaque
en réduisant la vasoconstriction provoquée par l’angiotensine II.
Si l’utilisation des IEC
au cours de l’insuffi-
sance cardiaque chronique, en complément du traitement clas-
sique, est actuellement incontournable, il n’en demeure pas moins
que la mortalité de cette affection reste élevée. Ainsi, au cours de
l’insuffisance cardiaque sévère, classe IV de la classification
NYHA, la mortalité, bien que diminuée sous IEC, reste sévère,
voisine de 36 % à un an (2). Quand l’insuffisance cardiaque est
moins grave, et correspond aux classes II et III de la classifica-
tion NYHA, la mortalité après 4 ans de traitement IEC avoisine
35 % (3). Il est donc apparu nécessaire de tenter d’améliorer ces
résultats ; de là les essais d’intervention sur le système adréner-
gique, mais aussi la réévaluation de l’action des IEC sur le système
rénine-angiotensine-aldostérone.
On a remarqué, au cours du traitement par IEC, que l’aldostérone,
diminuée dans un premier temps, avait tendance à remonter après
quelques mois de traitement. Ainsi, on a montré, au cours de l’hy-
pertension artérielle, qu’au-delà du premier mois de traitement
par captopril, le taux plasmatique d’aldostérone, initialement
abaissé par le traitement, remontait progressivement, et, après un
an de traitement, dépassait sa valeur initiale (8). Par contre, le taux
d’angiotensine II restait abaissé, ce qui semble démontrer que le
mécanisme de la remontée de l’aldostérone ne passe pas par un
échappement du blocage de l’enzyme de conversion. D’autre part,
lors du traitement de l’hypertension artérielle par énalapril, on a
observé une remontée de l’aldostérone plasmatique, mais égale-
ment du taux d’angiotensine II (9). Un phénomène voisin a été
observé au cours du traitement de l’insuffisance cardiaque par le
captopril. Après un an de traitement, celui-ci obtient une baisse
de l’angiotensine II, sans baisse du taux d’aldostérone. En outre,
la baisse de l’aldostérone ne s’observe qu’en cas de taux de rénine
élevé avant la mise en œuvre du traitement. Avec le zofénopril
donné au stade aigu de l’infarctus du myocarde, on a observé en
ACTUALITÉS
L’ aldostérone au cours
de l’insuffisance cardiaque
chronique
Aldostérone et IEC
.../...

La Lettre du Cardiologue - n° 322 - décembre 1999
9
quelques heures une baisse de l’enzyme de conversion, mais aussi
une remontée du taux de l’aldostérone (10).
L’échappement de l’aldostérone au traitement par IEC semble
être dû au fait qu’elle ne dépend pas uniquement du système
rénine-angiotensine, mais que sa sécrétion est également condi-
tionnée par le taux d’ACTH, par la kaliémie, et par le facteur
natriurétique auriculaire.
Tous ces éléments concourent à démontrer que le traitement par
IEC ne suffit à bloquer de façon ni complète ni prolongée la sécré-
tion potentiellement délétère de l’aldostérone.
Parce qu’elle se lie aux
mêmes récepteurs que
l’aldostérone, la spirono-
lactone est un antagoniste spécifique de ce minéralocorticoïde.
De ce fait, elle empêche la réabsorption du sodium et de l’eau au
niveau des cellules épithéliales des néphrons distaux, et évite la
fuite du potassium et du magnésium. Elle antagonise également
l’aldostérone au niveau de récepteurs d’autres tissus, en particu-
lier l’intestin grêle et les glandes salivaires. Grâce à l’ensemble
de ses effets, la spironolactone a des propriétés légèrement diu-
rétiques, et elle diminue le risque d’arythmies.
Sur des modèles expérimentaux, de faibles doses de spironolac-
tone préviennent la fibrose myocardique et des doses plus fortes,
antihypertensives, préviennent à la fois fibrose et hypertrophie
myocardiques (11).
Chez des insuffisants cardiaques, la spironolactone a montré des
vertus parasympathomimétiques. Elle a augmenté la variabilité
du rythme cardiaque, diminué la fréquence cardiaque, toutes
actions qui diminuent le risque d’arythmies. Elle a d’autre part
diminué le taux de l’aminopeptide précurseur du collagène, ce
qui suggère une réduction du renouvellement du collagène (6).
À l’heure actuelle, les
IEC sont la base du trai-
tement de l’insuffisance
cardiaque, mais ils combattent incomplètement l’hyperaldosté-
ronisme qui s’observe souvent, en particulier au cours de l’utili-
sation de diurétiques. Le problème s’est posé de savoir si la spi-
ronolactone pouvait être associée aux IEC, et quel était le risque
d’induire une hyperkaliémie dangereuse en associant deux pro-
duits qui favorisent la rétention de potassium.
Une étude prospective et randomisée, l’étude RALES, a eu pour
but de déterminer chez des insuffisants cardiaques la dose de spi-
ronolactone qui pouvait être associée au traitement par IEC, diu-
rétique et digoxine (12). Quatre doses ont été testées : 12,5, 25,
50 et 75 mg, contre un placebo, chez des insuffisants cardiaques
en classe II à IV de la NYHA, avec une fraction d’éjection abais-
sée en dessous de 40 %. Avec toutes les doses de spironolactone,
une baisse significative du peptide pro-atrial natriurétique auri-
culaire a été observée, de même qu’une augmentation de la rénine
plasmatique et de l’élimination urinaire d’aldostérone. Ces modi-
fications étaient dépendantes de la dose de spironolactone. La fré-
quence de l’hyperkaliémie (5,5 mmol/l) a augmenté avec les
doses de 50 et 75 mg de spironolactone. D’autre part, cette hyper-
kaliémie était significativement plus fréquente quand l’IEC uti-
lisé n’était pas du captopril, quand il existait à l’état basal une
kaliémie supérieure à 4,2 mmol/l, ou une créatininémie supé-
rieure à 16 mg/l (13). De plus, la dose d’IEC était un facteur pré-
dictif indépendant de l’hyperkaliémie. Les doses d’IEC utilisées
étaient en moyenne de 62 mg/j pour le captopril, de 14 mg/j pour
l’énalapril, et donc étaient inférieures aux doses recommandées.
Les recommandations qui résultent de cette première étude
RALES varient selon la gravité de l’insuffisance cardiaque. Si la
spironolactone est utilisée à titre prophylactique, une dose de
25 mg par jour est recommandée, tout en surveillant la kaliémie.
Si, par contre, il existe une insuffisance cardiaque sévère et réfrac-
taire, des doses plus importantes, jusqu’à 200 mg/j, peuvent être
utilisées transitoirement, sous étroite surveillance de la kaliémie.
Toute aggravation de l’insuffisance cardiaque doit conduire à une
vérification de la kaliémie, de crainte qu’une détérioration de la
fonction rénale conduise à une hyperkaliémie. Quand l’amélio-
ration des signes d’insuffisance cardiaque est obtenue, la dose de
spironolactone doit être diminuée (13).
Une autre étude contrôlée a montré, sur un plus petit nombre de
patients (n = 42), que l’adjonction de spironolactone au traite-
ment par IEC et diurétique avait des effets bénéfiques sur les
troubles du rythme (14). Ainsi, l’enregistrement holter a montré
une réduction significative des extrasystoles ventriculaires, pro-
portionnellement à l’augmentation du magnésium plasmatique.
Cette relation, qui n’est pas observée dans les suites d’un infarc-
tus, pourrait traduire la responsabilité des diurétiques dans la
déplétion en magnésium observée en cas d’insuffisance cardiaque
chronique (14). De tels effets suggèrent une possibilité d’amé-
liorer la survie des insuffisants cardiaques chroniques sous l’in-
fluence de l’inhibition de l’aldostérone. Cela explique la mise en
œuvre d’une seconde étude RALES, à la recherche d’un effet
favorable sur le pronostic de ces malades.
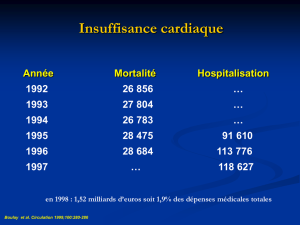
La seconde étude RALES a été menée chez plus de 1 600 insuf-
fisants cardiaques ayant une dysfonction ventriculaire gauche sys-
tolique attestée par une fraction d’éjection inférieure ou égale à
35 %, en classe NYHA III ou IV. Le but était de rechercher un
effet positif de la spironolactone sur la survie et sur l’évolution
de l’insuffisance cardiaque. Cette étude a été arrêtée en août 1998,
en raison de la constatation d’une amélioration très significative
des signes fonctionnels et de la survie (30 %, p < 0,0001) chez
les malades traités par spironolactone. La réduction de la morta-
lité concerne aussi bien les décès par insuffisance cardiaque que
les morts subites. La publication complète des résultats est toute
récente et montre que les bénéfices théoriques d’une inhibition
de l’aldostérone se traduisent en pratique clinique par une amé-
lioration significative de l’évolution de l’insuffisance cardiaque
chronique (15). C’est une preuve supplémentaire de la validité
de la théorie neurohormonale, précédemment confortée par l’ef-
ficacité des IEC et de certains bêtabloquants. ■
ACTUALITÉS
Les effets
de la spironolactone
Les essais d’association
de la spironolactone
aux IEC

La Lettre du Cardiologue - n° 322 - décembre 1999
10
RÉFÉRENCES BIBLIOGRAPHIQUES
1. Ho K.K., Pinsky J.L., Kannel W.B., Levy D. The epidemiology of heart failure :
the Framingham study. J Am Coll Cardiol 1993 ; 22 (suppl. A) : 6A-13A.
2. The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe
congestive heart failure. Results of the Cooperative North Scandinavian
Enalapril Study (CONSENSUS). N Engl J Med 1987 ; 316 : 1429-35.
3. The SOLVD Investigators. Effect of enalapril on survival in patients with redu-
ced left ventricular ejection fractions and congestive heart failure. N Engl J Med
1991 ; 325 : 293-302.
4. Packer M. The neurohormonal hypothesis : a theory to explain the mechanism
of disease progression in heart failure. J Am Coll Cardiol 1992 ; 20 : 248-54.
5. Swedberg K., Eneroth P., Kjekshus J., Wilhelmsen L. for the CONSENSUS
Trial Study Group. Hormones regulating cardiovascular function in patients with
severe congestive heart failure and their relation to mortality. Circulation 1990 ;
82 : 1730-6.
6. MacFadyen R.J., Barr C.S., Struthers A.D. Aldosterone blockade reduces vas-
cular collagen turnover, improves heart rate variability and reduces early mor-
ning rise in heart rate in heart failure patients. Cardiovasc Res 1997 ; 35 : 30-4.
7. Bauwens F.R., Duprez D.A., De Buyzere M.L. et coll. Influence of the arterial
blood pressure and nonhemodynamic factors on left ventricular hypertrophy in
moderate essential hypertension. Am J Cardiol 1991 ; 68 : 925-9.
8. Staessen J., Lijnen P., Fagard R., Verschueren L.J., Amery A. Rise in plasma
concentration of aldosterone during long-term angiotensin II suppression. J
Endocr 1981 ; 91 : 457-65.
9. Biollaz J., Brunner H.R., Gavras I., Waeber B., Gavras H. Antihypertensive
therapy with MK421 : angiotensin II-renin relathionships to evaluate efficacy of
converting enzyme blockade. J Cardiovasc Pharmacol 1982 ; 4 : 966-72.
10. Borghi C., Boschi S., Ambrosioni E., Melandry G., Branzi A., Magnani B.
Evidence of a partial escape of renin-angiotensin-aldosterone blockade in
patients with acute myocardial infarction treated with ACE inhibitors. J Clin
Pharmacol 1993 ; 33 : 40-5.
11. Weber K.T., Brilla C.G. Pathological hypertrophy and cardiac interstitium
fibrosis and renin-angiotensin-aldosterone system. Circulation 1991 ; 83 : 1849-65.
12. Pitt B. ACE inhibitor co-therapy in patients with heart failure : rationale for
the Randomized Aldactone Evaluation Study (RALES). Eur Heart J 1995 ; 16
(suppl. N) : 107-10.
13. The RALES Investigators. Effectiveness of spironolactone added to an angio-
tensin-converting enzyme inhibitor and a loop diuretic for severe chronic conges-
tive heart failure [the Randomized Aldactone Evaluation Study ( RALES)]. Am J
Cardiol 1996 ; 78 : 902-7.
14. Barr C.S., Lang C.C., Hanson J., Arnott M., Kennedy N., Struthers A.D.
Effects of adding spironolactone to an angiotensin-converting enzyme inhibitor in
chronic congestive heart failure secondary to coronary artery disease. Am J
Cardiol 1995 ; 76 : 1259-65.
15. Pitt B., Zannad F., Remme W.J. et coll. for the Randomized Aldactone
Evaluation Study Investigators. The effect of spironolactone on morbidity and
mortality in patients with severe heart failure. N Engl J Med 1999 ; 341 : 709-17.
ACTUALITÉS
1
/
5
100%