Lire l'article complet

La Lettre de l’Infectiologue • Tome XXIII - n° 5 - septembre-octobre 2008 | 175
MISE AU POINT
Interactions avec les nouveaux
antirétroviraux
Drug-drug interactions with new antiretroviral drugs
Lauriane Goldwirt*, Anne-Marie Taburet*
* Pharmacie clinique, CHU de Bicêtre,
AP-HP, Le Kremlin-Bicêtre.
D
arunavir (Prézista
®
), maraviroc (Celsentri
®
),
étravirine (ATU de cohorte) et raltégravir
(Isentress
®
) sont de nouveaux médicaments
antirétroviraux (ARV) puissants dont l’efficacité a
été démontrée en association avec un traitement
optimisé chez les patients lourdement prétraités
en échec virologique (1-5). Leur puissance d’action
repose soit sur un mécanisme d’action nouveau
impliquant de nouvelles cibles (maraviroc, anta-
goniste des récepteurs CCR5, ou raltégravir, inhi-
biteur de l’intégrase), soit sur une amélioration
de certaines caractéristiques virologiques, phar-
macocinétiques ou du profil de tolérance au sein
d’une classe (étravirine, nouvel INNTI, et darunavir,
nouvel inhibiteur de protéase). La mise en place
d’un traitement optimisé impose la maîtrise ou du
moins l’anticipation des éventuelles interactions
médicamenteuses (6). Seules les interactions d’ordre
pharmacocinétique seront évoquées ici. En effet, les
interactions d’ordre pharmacodynamique sont plus
faciles à prévoir, car elles relèvent le plus souvent
de la potentialisation d’effets thérapeutiques ou
d’effets indésirables connus.
L’étude pharmacocinétique des interactions est
réalisée en combinant deux types d’analyse. D’une
part, des études spécifiques évaluent la cinétique des
molécules seules et en association chez les adultes
volontaires sains randomisés en plusieurs groupes. Le
choix spécifique de l’association de molécules repose
sur la connaissance de leurs caractéristiques pharma-
codynamiques et pharmacocinétiques respectives,
sources d’interaction éventuelle, ou sur l’intérêt
thérapeutique que peut représenter leur coadmi-
nistration. Pour les associations thérapeutiques les
plus fréquentes, ces études figurent souvent dans
le dossier déposé par les industriels dans le cadre
de la demande d’AMM auprès de la Food and Drug
Administration (FDA) aux États-Unis ou de l’Euro-
pean Medicines Agency (EMEA) en Europe. D’autre
part, des études pharmacocinétiques de populations
réalisées au décours d’essais cliniques de phase III
permettent d’identifier les sources de variabilité
pharmacocinétique de la molécule testée, dont les
interactions pharmacocinétiques. Les résultats de
ces études permettent, le cas échéant, de classer les
interactions en fonction du risque prévisible pour le
malade, selon quatre niveaux de contrainte : “contre-
indication”, “association déconseillée”, “à prendre
en compte” ou “précaution d’emploi”.
Les interactions médicamenteuses peuvent en
théorie avoir lieu à toutes les étapes pharmaco-
cinétiques de la molécule (absorption, distribu-
tion, métabolisme ou excrétion), mais ce sont le
plus souvent l’absorption et le métabolisme qui
sont impliqués. En effet, les interactions au niveau
de l’absorption peuvent avoir pour origine des méca-
nismes physico-chimiques (adsorption ou dimi-
nution de l’absorption d’un médicament par un
antiacide, par exemple) ou des modifications de
l’effet de premier passage intestinal ou hépatique
impliquant les enzymes du métabolisme, dont l’acti-
vité peut être modifiée par des médicaments déclen-
chant une induction ou une inhibition enzymatique.
En fonction des caractéristiques pharmacocinétiques
des médicaments associés, les conséquences seront
une modification de la vitesse d’élimination par
métabolisme hépatique, et donc de la clairance,
ou une modification de la biodisponibilité. Certains
substrats, inducteurs enzymatiques, sont capables
d’augmenter la synthèse protéique de plusieurs
isoformes de cytochromes (phase I) et des enzymes
de phase II (glucuronoconjugaison par l’UGT, par
exemple) de manière non spécifique. L’inhibition est
le plus souvent compétitive entre deux substrats
d’une même enzyme (le plus souvent le cytochrome
P450, ou CYP), et donc plus spécifique. Les consé-
quences générales de ces interactions sont résumées
dans le tableau I, p. 178.

178 | La Lettre de l’Infectiologue • Tome XXIII - n° 5 - septembre-octobre 2008
Résumé
Quatre nouveaux antirétroviraux ont été mis sur le marché ces deux dernières années. Cet article fait le point sur
leurs interactions médicamenteuses avec d’autres antirétroviraux ou d’autres médicaments avec lesquels ils vont être
fréquemment associés. Le darunavir, un nouvel inhibiteur de protéase, possède les mêmes interactions que le ritonavir,
avec lequel il est systématiquement associé pour en augmenter les concentrations. Les conséquences cliniques de l’effet
d’induction enzymatique modeste de l’étravirine, un nouvel inhibiteur non nucléosidique de la transcriptase inverse, sont
probablement peu importantes. Le maraviroc, inhibiteur du récepteur CCR5, est un substrat du CYP 3A, et sa posologie
dépend des médicaments inhibiteurs du CYP (par exemple IP associé au ritonavir à faible dose) ou inducteurs (rifampicine)
auxquels il est associé. Enfin, le raltégravir, premier représentant de la classe des inhibiteurs de l’intégrase, est éliminé
par glucuronoconjugaison, et les interactions sont donc peu nombreuses et limitées à son association à un inhibiteur
de l’UGT 1A1, l’atazanavir, qui en augmente les concentrations d’environ 30 %, ou à des inducteurs puissants tels que
la rifampicine, qui en diminue les concentrations de 70 %.
Mots-clés
Darunavir
Raltégravir
Étravirine
Maraviroc
Interactions
Highlights
New antiretroviral drugs have
been approved recently. Daru-
navir as all other PIs is rito-
navir boosted and therefore
is a potent CYP 3A inhibitor.
Etravirine is a new NNRTI with
moderate enzyme induction
properties. Maraviroc is a CCR5
antagonist which is metabo-
lised by CYP 3A and the dose
to be administered is depen-
dent on whether it is combined
with a CYP 3A inhibitor (for
example ritonavir boosted PI)
or a potent enzyme inductor
such as rifampin. Raltegravir
is the first approved integrase
inhibitor. It is eliminated via
glucuronidation and therefore
potent drug-drug interactions
are limited to its combination
with atazanavir, an inhibitor
of UGT 1A1 leading to a 30%
increase in raltegravir concen-
trations or with potent enzyme
inducer such as rifampin, which
decreases raltegravir concen-
trations by 70%.
Keywords
Darunavir
Raltegravir
Etravirine
Maraviroc
Drug-drug interactions
Les interactions peuvent avoir des conséquences
cliniques délétères en majorant les effets indésirables
concentration-dépendants, dans le cadre d’une asso-
ciation à un inhibiteur (7), ou en induisant un risque
d’inefficacité thérapeutique, lors de l’association à un
inducteur enzymatique. Le ritonavir est un des exem-
ples emblématiques d’interaction favorable : inhibiteur
puissant du CYP 3A4, il est associé aux inhibiteurs de
protéase (IP) dont les caractéristiques pharmacociné-
tiques sont peu favorables (effet de premier passage
intestinal ou hépatique et demi-vie courte), pour en
augmenter l’exposition et donc l’efficacité ou en dimi-
nuer le nombre de prises quotidiennes.
La prise en charge thérapeutique du patient infecté
par le VIH est compliquée, car elle doit conjuguer
plusieurs traitements :
– le traitement antirétroviral, optimal, qui comprend
souvent au moins trois molécules, pour diminuer
la multiplication virale et éviter la sélection de
mutants résistants ;
– le traitement curatif ou préventif des infections
virales, bactériennes et fongiques opportunistes
liées à l’immunosuppression ;
– les traitements palliatifs des atteintes iatrogènes
liées à ces traitements ;
– le traitement des autres pathologies non liées au
VIH mais, par exemple, au vieillissement.
La bonne connaissance des profils métaboliques et
des interactions susceptibles de survenir avec les anti-
rétroviraux doit permettre de sélectionner les molé-
cules qui, au sein d’un traitement, minimiseront les
interactions et les effets indésirables et permettront
d’optimiser leur efficacité. Les interactions avec les
antirétroviraux commercialisés sont résumées dans les
rapports d’experts les plus récents ou ont fait l’objet
d’articles généraux (8, 9). Nous nous limiterons aux
interactions avec les quatre nouveaux antirétroviraux :
le darunavir associé au ritonavir, le raltégravir, le mara-
viroc et l’étravirine (en ATU de cohorte). À ce jour, ces
quatre nouveaux antirétroviraux disponibles sont indi-
qués chez les patients en échec. Chez ces patients,
le traitement comprendra au moins deux molécules
actives, appartenant à deux classes différentes, sélec-
tionnées en fonction du profil génotypique des virus.
C’est également chez ces patients qu’il conviendra de
prévenir, voire de guérir, les infections opportunistes
associées. La connaissance des interactions potentielles,
favorables ou défavorables, revêt donc une importance
considérable pour l’optimisation du traitement.
Caractéristiques
pharmacocinétiques
des nouveaux antirétroviraux
Les caractéristiques pharmacocinétiques sont
présentées dans le tableau II.
Darunavir
Le darunavir est un IP récent dont la puissance d’ac-
tion antirétrovirale repose sur la formation d’un
complexe stable avec la protéase virale de souches
sauvages ou mutées du VIH (10), ce qui lui confère
une barrière génétique très élevée. Administré per
os en deux prises quotidiennes, associé au ritonavir
à faibles doses (600 mg/100 mg), le darunavir est
rapidement absorbé (T
max
: 2,5 à 4 h) et présente
une biodisponibilité de 83 % (versus 37 % sans rito-
Tableau I. Conséquences de l’effet des inducteurs et inhibiteurs des enzymes du métabolisme des médicaments.
Inducteurs Inhibiteurs
Enzymes modifiées CYP, UGT, etc. CYP
Mécanisme Synthèse CYP Compétition ou fixation
Spécificité de l’effet Faible Importante
Délai d’action Maximal en 6-10 j Immédiat
Conséquences pharmacocinétiques Concentrations
Métabolites
Clairance
Concentrations
Métabolites
Clairance
Efficacité thérapeutique, toxicité

La Lettre de l’Infectiologue • Tome XXIII - n° 5 - septembre-octobre 2008 | 179
MISE AU POINT
navir) [11, 12]. Comme tous les IP, le darunavir est
substrat et inhibiteur de la P-glycoprotéine (PgP). Il
est lié aux protéines plasmatiques, essentiellement
à l’α-1-glycoprotéine (AGP) ; leurs interactions éven-
tuelles ont rarement des conséquences cliniques (13).
Le darunavir est métabolisé par le CYP 3A4 ; c’est
également un inhibiteur puissant de cette enzyme,
surtout associé au ritonavir, et à un moindre degré
des autres isoformes (2C9, 2C19, 2D6). Il semblerait
par ailleurs que le darunavir présente une activité
inductrice sur certains CYP.
Maraviroc
Le maraviroc est un inhibiteur d’entrée, antagoniste puis-
sant des récepteurs CCR5, indiqué dans le traitement en
association des patients VIH colonisés exclusivement
par des souches à tropisme CCR5. La posologie est de
300 mg x 2/j, en l’absence d’interaction médicamen-
teuse. L’absorption du maraviroc est très variable (T
max
de 2 à 4 h), avec une biodisponibilité de 25 à 35 % (14).
Cette variabilité est probablement liée à un mécanisme
saturable d’efflux par transporteurs intestinaux. Substrat
de la PgP et du CYP 3A4 (voie majoritaire du métabo-
lisme), le maraviroc est également éliminé par sécré-
tion tubulaire rénale (25 % de la dose éliminée par les
urines sous forme inchangée). Le potentiel d’interaction
en association avec d’autres antirétroviraux est donc
élevé. D’autre part, bien qu’aucune marge thérapeu-
tique efficace n’ait été définie, des effets indésirables
sévères (allongement de QT, hypotension orthostatique,
atteinte hépatique) ont été rapportés au cours d’es-
sais de phase III, dont la survenue a été associée à une
concentration plasmatique élevée de maraviroc.
Raltégravir
Le raltégravir est un inhibiteur de l’intégrase du VIH-1
dont la puissance et la rapidité de la réponse virolo-
gique s’accompagnent d’une faible barrière géné-
tique. Il est indiqué en association à la posologie de
400 mg x 2/j chez le patient VIH lourdement prétraité
résistant à plusieurs classes d’antirétroviraux. Il est
rapidement absorbé et est éliminé de 7 à 14 % par
voie rénale sous forme inchangée (5). Le raltégravir est
métabolisé par glucuronoconjugaison par l’UGT 1A1
(majoritairement), et c’est un substrat de la PgP (15).
La solubilité du raltégravir étant fonction du pH, les
variations du pH gastrique ont un effet direct sur
l’absorption. Aucun effet inhibiteur ni inducteur du
CYP 3A4 n’a été démontré. Aucun effet indésirable
concentration-dépendant n’a été rapporté au cours
des essais cliniques. Par ailleurs, aucune corrélation
entre la concentration et l’efficacité virologique n’a
été établie, la seule donnée disponible étant l’IC
90
,
estimée in vitro à 33 nM.
Étravirine
L’étravirine est un INNTI du VIH-1. Administré en deux
prises per os de 200 mg/j, l’étravirine a démontré
son efficacité virologique puissante chez les patients
VIH-1 lourdement prétraités en échec virologique.
L’ATU de cohorte de ce médicament inclut la condition
de l’associer au moins à un inhibiteur de protéase.
L’étravirine est un substrat et faible inducteur des
CYP 3A4, 2C9, un substrat du CYP 2C19, et est faible-
ment inducteur de la PgP. Il est également métabolisé
par l’UDPGT, enzyme qu’il induit. La demi-vie d’élimi-
nation de l’étravirine est de 30 à 40 heures.
Interactions entre
médicaments antirétroviraux
La prise en charge optimale du patient infecté par le
VIH comprend l’association d’au moins trois médica-
ments antirétroviraux (9). Connaître les associations
favorables ou délétères revêt donc une importance
Tableau II. Résumé des caractéristiques pharmacocinétiques des nouveaux antirétroviraux.
F (%) Effet repas fu (%) fe (%) Métabolisme Effet inducteur/inhibiteur
Darunavir/
ritonavir
ND R 94 < 5 CYP 3A Inhibition du CYP 3A4 +++
Induction de CYP + et UGT
Étravirine ND ND 99,9 < 1 CYP 3A
CYP 2C
Induction de CYP
Induction de l’UGT
Raltégravir ND R 83 < 5 UGT 1A1 Aucun
Maraviroc 25-35 S 76 25 CYP 3A Aucun
F : biodisponibilité ; fu : fraction libre, non fixée aux protéines plasmatiques ; fe : fraction de la dose administrée excrétée dans les
urines sous forme inchangée ; ND : non disponible ; R : à prendre avec un repas ; CYP : cytochrome P450 ; UGT : UDP-glucuronosyl-
transférase ; S : sans effet repos.

180 | La Lettre de l’Infectiologue • Tome XXIII - n° 5 - septembre-octobre 2008
Interactions avec les nouveaux antirétroviraux
MISE AU POINT
majeure, d’autant que les nouveaux traitements
vont être associés le plus souvent chez les patients
en échec virologique et que leur efficacité n’a pas
toujours fait l’objet d’une évaluation immunovirolo-
gique dans le cadre d’essais cliniques. Les interactions
ayant fait l’objet d’études chez les volontaires sains
ou chez des patients dans le cadre d’essais cliniques
sont résumées dans le tableau III.
Les essais DUET 1 et 2 ont permis d’évaluer l’effi-
cacité et la tolérance de l’association darunavir/
ritonavir et étravirine (1, 2). L’essai ANRS139 (TRIO),
actuellement en cours, évalue l’efficacité de l’asso-
ciation du raltégravir, du darunavir et de l’étravirine.
Il permettra de déterminer l’effet de l’étravirine et
des inducteurs modérés que sont le darunavir/rito-
navir, sur les concentrations de raltégravir.
Darunavir
Le darunavir est, comme tous les IP, associé au rito-
navir à faible dose (100 mg x 2/j) afin d’en augmenter
l’exposition. Il n’a pas été démontré d’intérêt virolo-
gique ni clinique à associer deux IP. La coadministra-
tion de tipranavir et de darunavir pourrait présenter
un intérêt théorique chez certains patients, en raison
de leur profil de résistance distinct et de la suscepti-
bilité différente des souches qui peuvent co-infecter
un patient (16). Il est cependant très probable que le
tipranavir diminuera les concentrations de darunavir,
comme celles d’autres IP associés (17).
Bien que le ténofovir soit éliminé par sécrétion rénale
stricte et qu’il n’interagisse pas avec le CYP 3A4
ni en tant que substrat ni en tant qu’inducteur ou
inhibiteur, son association au darunavir conduit, chez
l’adulte sain, à une augmentation de l’exposition
aux deux molécules. Le mécanisme de l’interaction
impliquerait une augmentation de la perméabilité
intestinale au ténofovir sous l’action du darunavir
(effet commun aux autres IP) [18] et/ou un effet
inhibiteur du ritonavir sur les transporteurs MDR-1
rénaux.
Les essais cliniques DUET 1 et 2 ont évalué l’effica-
cité de l’étravirine versus placebo chez les patients
VIH multirésistants et dont le traitement opti-
misé comporte du darunavir associé au ritonavir à
faible dose (2, 3). Il a été démontré que le darunavir
(ou, plus probablement, le ritonavir associé) diminue
les concentrations d’étravirine (réduction de l’aire
sous la courbe [ASC] de l’étravirine de 30 %), ce
qui, compte tenu des résultats de l’essai, n’a pas de
conséquences cliniques (19).
Étravirine
Par un mécanisme inconnu, l’étravirine augmente les
concentrations de darunavir de 23 % (essais DUET 1
et 2) [2, 3]. Les IP associés au ritonavir diminuent
les concentrations d’étravirine (19). À l’inverse,
l’étravirine diminue les concentrations de saqui-
navir administré sans ritonavir (réduction de l’ASC
Tableau III. Interactions d’ordre pharmacocinétique entre médicaments antirétroviraux.
Les variations de concentration sont exprimées en variation de l’ASC par rapport à chaque médicament administré seul.
IP NNRTI INRT Inhibiteurs d’entrée
Inhibiteurs d’intégrase
Darunavir/antirétroviral (DRV) IDV 23 %
LPV/RTV 37 %
SQV ↔
DRV 24 %
DRV 53 %
DRV
EFV 21 %
NVP 27 %
DRV 13 %
DRV 24 %
TDF 22 % DRV 21 % MRV 395 % DRV ↔
Maraviroc (MRV) SQV/RTV ↔
LPV/RTV ↔
ATV ↔
ATV/RTV ↔
DRV/RTV ↔
RTV ↔
MRV 877 %
MRV 283 %
MRV 257 %
MRV 388 %
MRV 395 %
MRV 161 %
EFV ↔
LPV/RTV + EFV
SQV + EFV
ETV 6 %
MRV 45 %
MRV 153 %
MRV 400 %
MRV 53 %
Raltégravir (RTG) ATV ↔
ATV/RTV ↔
TPV/RTV ↔
RTG 95 %
RTG 41 %
RTG 24 %
EFV ↔
ETV 10 %
RTG 36 %
RTG 10 %
TDV ↔RTG 49 %
Étravirine (ETV) ATV 14 %
IDV 46 %
SQV ↔
LPV/RTV 19 %
APV 65 %
TPV 18 %
DRV 13 %
ETV 30 %
ETV 51 %
ETV 37 %
ETV 17 %
ETV 76 %
EFV 21 %
EFV ND
NVP ND
ETV 41 %
ETV 55 %
TDV 15 %
DDI ↔
ETV 19 %
ETV 11 %
MRV 53 %
RTG 10 %
ETV 6 %
ETV 10 %

La Lettre de l’Infectiologue • Tome XXIII - n° 5 - septembre-octobre 2008 | 181
MISE AU POINT
de 52 %), mais l’exposition du saquinavir associé
au ritonavir n’est pas modifiée en présence d’étra-
virine. En présence d’étravirine, les concentrations
de lopinavir restent inchangées (20, 21), mais celles
d’amprénavir augmentent de 69 % (22). Enfin, l’asso-
ciation étravirine/atazanavir n’a pas été évaluée.
Lorsque l’étravirine est associé au raltégravir, les
concentrations d’étravirine restent inchangées. En
revanche, l’effet inducteur modeste de l’étravirine sur
l’UGT se traduit par une diminution des concentra-
tions de raltégravir dont les conséquences cliniques
sont inconnues (23).
L’éfavirenz et la névirapine diminuent très significa-
tivement la concentration plasmatique d’étravirine
(ASC de l’étravirine diminuée de 41 % et 55 %
respectivement) [24]. Le mécanisme impliqué est
probablement l’induction du CYP 3A4 par les INNTI.
Ces associations ne sont pas recommandées sur
le plan pharmacocinétique et, d’autre part, sur les
plans pharmacodynamique et virologique, elles ne
sont pas pertinentes.
Maraviroc
L’association du maraviroc à un inhibiteur de CYP
(IP sans activité inductrice de CYP) conduit à une
majoration de l’ASC du maraviroc allant de + 161 %
pour le ritonavir à + 877 % pour le saquinavir boosté
(25). L’association au tipranavir n’entraîne pas
d’interaction cliniquement significative, probable-
ment parce que les effets inhibiteurs et inducteurs
se compensent (26). La dose de maraviroc recom-
mandée en cas de coadministration avec les IP (tipra-
navir exclu) est réduite de moitié, soit 150 mg x 2/j,
et ce, quelle que soit l’ampleur de la surexposition
au maraviroc constatée chez le volontaire sain. La
coadministration du maraviroc avec des inducteurs
stricts du CYP 3A4, tels que l’éfavirenz, s’accompagne
d’une réduction significative de l’ASC du maraviroc
(27). Bien que l’impact clinique de cette diminu-
tion de l’exposition ne soit pas clairement défini
(absence de marge thérapeutique à ce jour), il est
recommandé de doubler la dose de maraviroc, soit
600 mg x 2/j. En l’absence d’évaluation de l’efficacité
virologique et de la tolérance à long terme de telles
associations, leur utilisation doit être prudente
et faire l’objet d’un suivi immunovirologique et
clinique renforcé. En l’état actuel des connaissances,
un suivi thérapeutique pharmacologique ne peut
être recommandé. La mise en place d’études serait
nécessaire afin d’individualiser au mieux la posologie
du maraviroc selon les antirétroviraux associés.
Raltégravir
Le raltégravir n’étant pas substrat du CYP, mais de
l’UGT, les interactions sont plus rares. L’association
à l’éfavirenz entraîne une sous-exposition au ralté-
gravir (ASC diminuée de 36 %) ; le doublement de la
dose de raltégravir est alors recommandé. L’étravirine
diminue modérément les concentrations de ralté-
gravir ; il n’existe pas à ce jour de recommandation
posologique (23). À l’inverse, l’atazanavir est un
inhibiteur puissant de l’UGT 1A1, et augmente donc
très significativement l’ASC du raltégravir. Cette
augmentation de l’exposition au raltégravir ne
semble pas associée à un risque accru d’intolérance
ou de toxicité. Cependant, l’intérêt réel d’une telle
association reste à démontrer.
Interactions
des antirétroviraux
avec d’autres médicaments
Seules les interactions avec les médicaments à marge
thérapeutique étroite ou fréquemment associés aux
antirétroviraux seront évoquées. Il s’agit des statines,
des contraceptifs oraux et de la méthadone. Les
principales interactions décrites sont résumées dans
le tableau IV.
Statines
Hormis les antirétroviraux et les anti-infectieux,
d’autres classes médicamenteuses ont fait l’objet
d’études, notamment les inhibiteurs de la HMG CoA
réductase (statines), utilisés chez les patients VIH pour
pallier les perturbations du métabolisme lipidique
iatrogènes ou liées à l’infection VIH. À l’exception
de la fluvastatine (métabolisée par le CYP 2C8), de la
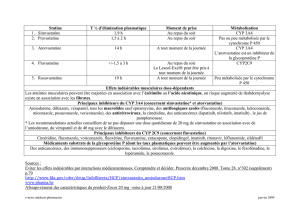
Tableau IV. Interactions des nouveaux antirétroviraux avec les autres médicaments.
Statines Contraceptifs oraux Méthadone
Darunavir Atorvastatine 85 %
Pravastatine (x 2 à 3 chez certains
patients)
Éthinylestradiol 44 %
Noréthindrone 14 %
Forme R– 16 %
Forme S– 36 %
Maraviroc ↔ ↔ ↔
Raltégravir ↔Éthinylestradiol ↔
Norgestimate 16 %
↔
Étravirine Atorvastatine 37 %
Métabolites actifs 27 %
Éthinylestradiol 22 %
Noréthindrone 5 %
↔
Forme S– 11 %
Forme R– 6 %)
 6
7
8
6
7
8
1
/
8
100%