Télécharger le book de l`AG

PERNETTA
SAKK 22/10 / UNICANCER UC-0140/1207
Coordonnateur : Pr Hervé BONNEFOI
Etude randomisée de phase II évaluant un traitement par le pertuzumab associé au
trastuzumab avec ou sans chimiothérapie, suivi du T-DM1 en cas de progression, chez des
patientes atteintes d’un cancer du sein métastatique HER2 positif.
METHODOLOGIE : étude de phase II ouverte, multicentrique, randomisée.
DESIGN :
OBJECTIF PRINCIPAL :
Comparaison du traitement sans chimiothérapie, composé du trastuzumab et du pertuzumab suivi
d’un traitement de deuxième ligne par T-DM1 en cas de progression de la maladie, avec une
chimiothérapie initiale combinée au trastuzumab et au pertuzumab suivie d’un traitement de deuxième
ligne par T-DM1 en cas de progression de la maladie.
OBJECTIF SECONDAIRES :
- Evaluation de l’efficacité
- Evaluation la sécurité
- Evaluation du profil de tolérance
- Evaluation de la qualité de vie
- Enregistrement des thérapies antitumorales ultérieures.
PRINCIPAUX CRITÈRES D'INCLUSION DE PREMIÈRE LIGNE DE TRAITEMENT :
Cancer du sein métastatique confirmé histologiquement.
Tumeur HER2 positive selon un test HER2 positif confirmé par la pathologie centrale.
Femmes ≥18 ans.
Statut de performance OMS des patientes 0 à 2.
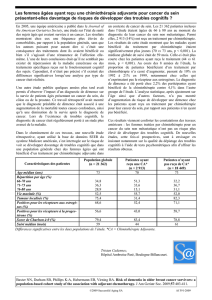
NOMBRE DE PATIENTES
ATTENDUES :
208 DONT 120 EN
FRANCE
En recrutement

Fraction d’éjection du ventricule gauche ≥50%, déterminée par échocardiographie ou
ventriculographie isotopique.
Fonctions organiques suffisantes.
PRINCIPAUX CRITÈRES DE NON-INCLUSION DE LA PREMIÈRE LIGNE :
Chimiothérapie préalable en cas de cancer du sein inopérable localement avancé.
Traitement antérieur anti- HER2 pour cancer du sein métastatique ou inopérable.
Plus d’une ligne thérapeutique endocrine contre un cancer du sein métastatique ou un cancer
du sein inopérable excédant une durée de 1 mois
Traitement antérieur avec pertuzumab et/ou T-DM1.
Métastases méningées ou du SNC connues
Métastase(s) osseuse(s) isolée(s) traitée(s) par radiothérapie (si la/les métastase(s)
osseuse(s) représente(nt) la/les seule(s) lésion(s) tumorale(s)).
Neuropathie périphérique de grade ≥2 (selon CTCAE).
Présence d’hypertension (systolique >160mmHg et/ou diastolique >100mmHg) ou angine de
poitrine, exigeant un traitement médicamenteux.
Antécédent d’insuffisance cardiaque chronique NYHA III +IV
Arythmies à risque élevé non contrôlées.
Infarctus dans les 6 mois précédant la participation à l'étude (randomisation).
PRINCIPAUX CRITERES D'INCLUSION POUR LA DEUXIEME LIGNE DE TRAITEMENT :
Au moins une dose de traitement de première ligne.
Progression de la patiente lors de la première ligne de traitement ou suite à une radiothérapie
de métastase osseuse.
Fonctions organiques suffisantes, déterminées par les paramètres biologiques similaires à la
première ligne:
Fraction d’éjection du ventricule gauche ≥50%, déterminée par échocardiographie ou
ventriculographie isotopique.
PRINCIPAUX CRITÈRES DE NON INCLUSION DE LA DEUXIÈME LIGNE DE TRAITEMENT :
Arrêt de la 1ère ligne de traitement dû à des toxicités intolérable sans preuves objectives de
la progression de la maladie.
Des métastases du système nerveux central non traitées nécessitant un traitement
symptomatique, une radiothérapie, une chirurgie ou un autre traitement, y compris les
stéroïdes, pour contrôler les symptômes dans les 2 mois (60 jours) avant la randomisation
Neuropathie périphérique de grade ≥ 3 CTCAE.
Maladie pulmonaire interstitielle (ILD) ou pneumopathie de grade ≥ 3 CTCAE.

COMET – GRT02
UC-0102/1203
Coordonnateur : Pr Jean-Yves PIERGA
Etude de cohorte de validation prospective de facteurs prédictifs biologiques et d’imagerie de la
réponse au bevacizumab (AVASTIN®) associé à une chimiothérapie par paclitaxel hebdomadaire
en 1ère ligne de traitement chez des patients atteints d’un cancer du sein métastatique.
MÉTHODOLOGIE: Étude de cohorte, multicentrique en ouvert à bras unique.
En recrutement
Blood samples:
ctDNA

OBJECTIF PRINCIPAL:
Valider prospectivement les taux initiaux et de variations des CEC/CTC (étude biologique) ainsi que la
graisse viscérale (étude imagerie) comme facteurs prédictifs de survie sans progression et de réponse
à l’association bevacizumab et paclitaxel, en première ligne de traitement du cancer du sein
métastatique.
OBJECTIF(S) SECONDAIRE(S) :
Valider prospectivement des facteurs prédictifs de survie sans progression, de survie globale et de
réponse à l’association bevacizumab et paclitaxel, en première ligne de traitement du cancer du sein
métastatique à partir d’études biologiques, pharmacogénétique et protéomique.
Evaluer la Qualité de vie
- Impact de la progression de la maladie et de la toxicité
- Rôle pronostique sur la survie globale
- Impact de la graisse viscérale
Corréler les marqueurs biologiques avec la tolérance au traitement.
PRINCIPAUX CRITÈRES D’INCLUSION :
Homme ou femme âgé(e) de 18 ans ou plus.
Adénocarcinome du sein confirmé
histologiquement, au stade métastatique
(lésion mesurable ou non mesurable), HER2
négatif (sur le dernier tissu tumoral analysé),
Patient(e) devant recevoir en première ligne
de chimiothérapie une association paclitaxel
hebdomadaire et bevacizumab selon les
recommandations de l’EMEA.
Statut des récepteurs hormonaux (RO et RP)
renseigné
Indice de performance ECOG ≤ 2.
Espérance de vie ≥ 12 semaines.
PRINCIPAUX CRITÈRES DE NON INCLUSION:
Chimiothérapie antérieure pour la maladie
métastatique ;
Hormonothérapie concomitante
Le(la) patient(e) ne doit pas avoir subi de
radiothérapie pour le traitement de la
maladie métastatique (à l'exception des cas
de radiothérapie à visée antalgique pour des
douleurs osseuses d'origine métastatique).
Hypersensibilité connue au paclitaxel et/ou
au bevacizumab ou à l'un des excipients.
NOMBRE DE PATIENTES
ATTENDUES:
510

RTS 02 – SHARE
UC-0140/1001
Coordonnateurs :
Pr Yazid BELKACEMI - Scientifique
Dr Eric LARTIGAU – Bonnes pratiques
Dr Céline BOURGIER – Contrôle qualité
Essai de phase III multicentrique comparant une irradiation accélérée focalisée au lit opératoire
à une irradiation mammaire standard ou hypofractionnée dans le cancer du sein à faible risque
de rechute locale.
MÉTHODOLOGIE : Etude prospective ouverte, contrôlée, multicentrique de phase III de non infériorité,
randomisée, comparant l’IPAS (traitement expérimental) à une irradiation mammaire en totalité selon un
schéma standard ou un schéma hypofractionné (traitements contrôles).
DESIGN :
OBJECTIF PRINCIPAL : Estimer et comparer les taux de rechutes intra-mammaires homolatérales entre le
traitement expérimental (IPAS : Irradiation Partielle et Accélérée du Sein) et les traitements contrôles
(irradiation mammaire en totalité selon un schéma standard ou un schéma hypofractionné).
OBJECTIF(S) SECONDAIRE(S)
EFFICACITÉ
o Evaluer et comparer dans le traitement expérimental et les traitements contrôles :
la survie sans récidive intra mammaire homolatérale
la survie sans récidive ganglionnaire
la survie sans récidive métastatique à distance
la survie sans cancer controlatéral
la survie spécifique
la survie globale
TOLÉRANCE
o Evaluer et comparer le taux et le type de toxicités (aiguës et tardives) dans le traitement
expérimental et les traitements contrôles
o Evaluer et comparer les résultats trophiques
ESTHÉTIQUE
o Evaluer et comparer les résultats esthétiques (patiente/praticien)
QUALITÉ DE VIE
o Mesure de la Qualité de vie et de la satisfaction patiente
MÉDICO-ÉCONOMIQUE
En recrutement
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
1
/
45
100%