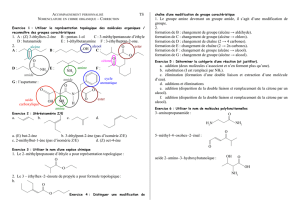
Résumé Chimie Générale

La théorie a savoir
Chapitre 1:
Il existe 3 types de liaison: Covalente, ionique , Covalente Polaire.
Les Carbones ont trois types d’hybridation, sp³ pour des liaisons simples, sp²
Pour les liaison double et sp pour les triples.
Les orbitales de recouvrement des liaisons multiples sont des liaisons .π
Nous pouvons séparer les composées en 2 Catégories, les Acides et les Bases.
Une Base donne une paire d’électrons ou alors accepte en H+, un Acide accepte une paire d’électrons ou
alors donne un H+.
Pour qu’une réaction soit équilibrée il faut que les réactifs soient des acides et bases plus fortes que les
produits.
On peut également connaître l’issue d’une réaction par le K1 il peu être calculer en divisant la concentration
du produit par la concentration des réactifs. Si le K est > 1 la réaction est favorable et si il est <1 c’est
l’inverse.
Chapitre 2 :
Un groupe fonctionnel est un groupe sur une molécule qui a une réactivité différentes (elle va en générale
prendre le dessus sur la réactivité de la molécule).
Les alcanes sont hydrocarbure plutôt inerte, ils sont dis saturés car ils ne possèdent pas de doubles liaisons.
Les isomères de constitution sont des composés aillant la même formule brute mais un arrangement
différent des atomes (Ex : Butane et isobutane).
Une rotation autour de la liaison c-c est possible dans les alcanes. Pour mieux la voir une projection de
Newman est nécessaire. La conformation décalé est bien plus stable que l’éclipsé. Mais ces deux
conformations s’échange très rapidement.
Pour les cycloalcanes la rotation n’est pas possible mais il existe tout de même une conformation propre au
cycloalcanes, ce sont des carbones Diastéréoisomères CIS/TRANS.
Les Cyclohexanes ont une conformation très stable, la conformation chaise, les substituants accrochée aux
cycles peuvent avoir une position axiale ou équatoriale indépendamment de la conformation CIS/TRANS.
Chapitre 3 :
Les Alcènes sont des hydrocarbures à doubles liaisons. Ils sont qualifiés de carbones insaturés.
Pour les alcènes la rotation autour de la liaison n’est pas possible alors il existe les isomères CIS/TRANS et
si les 4 substituants sont différents alors on a à faire a des énantiomères Z/E.
Une réaction peu être décrite schématiquement en utilisant un diagramme qui représente les énergies
nécessaires à cette réaction. (Énergie d’activation, intermédiaire carbocationique).
Chapitre 4:
Dans ce chapitre deux règles très importantes de chimie sont citées. La première est celle de Markovnikov,
elle dit que lors de l’addition d’un halogénoaclane (HX) sur un alcène, l atome d’hydrogène se fixera sur le
carbone le moins substitué et que l’halogène lui se fixera sur le carbone le plus substitué. La deuxième est
celle de Zaitsev qui dit que lors de l’élimination de l’eau ou d’un halogène l’alcène le plus substitué sera
formé.
Un nouveau composé est vu dans ce chapitre : Les diènes conjugués. Ils possèdent une alternance de
doubles liaisons, qui leur donne une configuration particulière, pour ces composés la règle de Markovnikov
n’est pas vraiment applicable, car lors de l’addition d’un composé un carbocation allylique avec une
charge a 2 endroit possible, le résultat sera 2 composés différents.
Les Alcynes suivent en générales les mêmes règles que les alcènes.
Les réactions :
Addition HX sur un alcène halogénoalcane (produit Trans et suit la règle de Markov.)→

Addition H2 sur un alcène alcane (produit Cis)→
Addition X2 sur un alcène dihalogénoalcane (produit Trans)→
Addition H2O sur un alcène alcool (suit la règle de Markov.)→
Oxydation d’un alcène par KMnO4 : en milieu basique Diol (produit Cis)→
en milieu acide Cétone (si le C n’a pas de H), →
Acide carbo. (si le C a 1 H)
CO2 (si le a 2H)
Polymérisation des alcènes → poly alcane.
Elimination de H2O sur un alcool tertiaire par H2SO4 alcène (règle de Zaitsev)→
Elimination de HX sur un halogénoalcane par une base forte alcène (règle de Zaitsev)→
Addition HX sur un alcyne halogénoalcène (règle de Markovnikov)→
Addition H2 sur un alcyne -traité avec Pd alcane→
-traité avec un catalyseur de Lindlar Alcène→
Addition X2 sur un alcyne dihalogénoalcène (produit Trans)→
Addition H2O sur un alcyne [énol] cétone→ →
Enrichissement d’un alcyne : création d’un anion (réagit avec NaNH2) puis réaction avec un
halogénoalcane.
Chapitre 5 :
Les benzènes sont des composés aromatiques relativement inertes. Lorsqu’ils sont substitué on leur attribue
un préfixe para, méta, ou ortho suivant la position des 2 substituants dans le cycle.
Lors d’une réaction avec un benzène (le composé est un électrophile), les électrons de la double liaison π
attaque le composé, un carbocation est formé jusqu'à ce le benzène lâche un protons et récupère par la même
occasion les électrons qu’il a utilisé.
Les substituants sur benzène peuvent affecter la réactivité ainsi que l‘orientation du prochain substituant.
Dans les méta désactivateurs : COOH. , C=OH, C=OCH3, NO2, SO3H, CN, NR3+.
Dans les ortho para désactivateurs : Br, I, F, Cl.
Dans les ortho para activateurs : OH, CH3, COCH3, NH2, NHCOCH3, et le benzène
Les réactions :
Bromation du benzène en présence de FeBr3 cycle-Br + HBr→
Chloration du benzène en présence de FeCl3 cycle-Cl + HCl→
Nitration du benzène : 1° HNO3 + H2SO4 NO2+ + H2O→
2° benzène + NO2+ cycle-NO2 →
Sulfonation du benzène : 1° SO3 + H2SO4 HSO3+ + HSO4-→
2°benzène + HSO3+ cycle-HSO3→
Alkylation de Friedel-Craft : Benzène + halogénoalcane cycle-R + HX→
Acylation de Friedel-Craft : Benzène + halogénure d’acyle cycle-cetone + HX→
Oxydation : cycle-R + KMnO4 cycle-COOH→
Réduction (Hydrogénation) (en présence de PtO2) cycloalcanes.→
Chapitre 6 :
Les carbones stéréogènes sont des carbones possédant quatre substituants différents, appelé aussi molécule
chirale ou énantiomère. Ces molécules ne possèdent pas de plan de symétrie. Les composées énantiomères
sont optiquement actives car lorsqu’ils se trouvent en solution et qu’ils sont traversés par une lumière
polarisée, ils dévient cette lumière d’un certain angle.
On peut différencié ces centres stéréogènes en R ou S si les trois substituants les plus importants tournent
vers la droite il sera R et sinon S.
Les doubles ou triples liaisons sont considéré comme des liaisons simples ou l’on rajoute l’atome
correspondant un 2ème ou 3ème fois. Ex : C=O C-C→-O-O.

Les diastéréoisomères sont des composés qui possèdent 2 centres stéréogènes. Mais par exemple si un
composé est R, R le composé S, S sera son énantiomère, tandis que celui qui sera R, S ou S, R sera un
diastéréoisomère (par rapport a R,R et S,S). Donc si les 2 centres sont opposés, ce sont des énantiomères
Et si au moins un centre est pareil et que le l’autre ne l est pas alors c’est un diastéréoisomère.
Les composés méso sont des composé qui ont au premier abord pourrai être des composés énantiomères
mais qui ne le sont pas car ils possèdent un plan de symétrie entre les 2 carbones stéréogènes.
Un mélange racémique est un mélange 50/50 de 2 énantiomères (activité optique nul).On peut les séparer
de ce mélange par chromatographie ou par dédoublement.
Chapitre 7 :
Les substitutions nucléophiles font selon deux mécanismes.
La première est Sn2 :
Dépend de la concentration des deux réactifs.
Attaque du nucléophile avec un angle de 180°
Inversion de configuration
N’est favorable que en présence de réactifs primaire ou secondaire à cause de l’encombrement
stérique.
La deuxième est Sn1 :
Dépend de la concentration du substrat.
Dissociation spontanée du substrat
Mélange de 2 configurations car il y a formation d’un carbocation et 2 attaques possible
N’est favorable qu’avec des produits tertiaires.
Les bon groupes partant : I Br H2O = Cl F.→ → →
Les réactions d’élimination se font aussi selon 2 mécanismes :
La première est E2 :
Lorsqu’une base arrache un proton ce qui provoque le départ d’un groupe partant
Elle a lieu selon une géométrie antiplanaire
Donne lieu a des alcènes E ou Z tout dépend de la position initial des substituants.
La deuxième est E1 :
Il y a dissociation spontanée du substrat
Formation d’un carbocation, qui perd un proton pour récupérer sa paire d’e-.
Favorable avec les composé tertiaires
Du point de vue des composés afin de déterminer à quel type de réaction nous avons à faire, nous pouvons
dire que :
Les composés primaire et secondaire réagissent selon Sn2 en présence d’un bon nucléophile, et
selon E2 en présence d’une base forte.
Un halogénoalcane tertiaire va réagir selon E2 en présence d’une base forte
Un alcool tertiaire va réagir selon E1 en présence de H2SO4, et selon Sn1 en présence de HBr ou
HI ect…
Les nucléophiles :
Iodure, Hydrure, Chlorure, méthanoate, méthane thiolate, triméthylamine, acétate, hydrogénosulfure,
hydroxyde, Ammoniac, cyanure, azoture.
Les réactions des halogénoalcane :
Déjà-vu : addition HX ou X2 sur un alcène
Synthèse des halogénoalcanes par chloration radicalaire
Réaction d’un alcool tertiaire avec HX halogénoalcane (selon SN1)→
Réaction d’un alcool secondaire ou primaire avec PBr3 ou SOCl2 halogénoalcane→

Réaction d’un halogénoalcane avec un réactif de Grignard inversion de polarité→
Chapitre 8 :
Les alcools et les phénols sont des composé similaire a l’eau ils ont la même géométrie et sont capable de
faire des pont hydrogènes et donc on des point d’ébullition plus élevé. Ces propriété ne sont pas applicable
aux éthers oxydes. Comme l’eau ils sont un peu acides et un peu basiques à la fois. Les phénols sont plus
acide car il peuvent stabilisé leur charge par résonance, mais cette acidité va aussi dépendre du substituant
qui sera sur le phénol, si c’est un électro-attracteur il augmentera l’acidité du phénol et il la baisse si c’est
un groupe électro-donneur.
Les réactions :
Synthèse des alcools :
oDéjà vu : Addition H2O sur un alcène et oxydation d’un alcène.
oAldéhyde en présence de NaBH4 alcool primaire→
oCétone en présence de NaBH4 alcool secondaire→
oAcide carbo. en présence LiAlH4 alcool primaire→
oEster en présence de LiAlH4 2 alcools→
oEther oxyde avec un HX alcool + RX→
Réaction des alcools :
oDéjà vu : transformation en halogénoalcanes, élimination de H2O
oAlcools primaires oxydé par PCC Aldéhyde→
oAlcools primaires oxydé par CrO3 Acide carbo.→
oAlcools secondaires oxydé par PCC Cétone→
Synthèse des éthers oxydes :
oRéaction de Williamson : alcool + Na alcoolate →
Alcoolate + RX éther oxyde→
Réaction des éthers oxydes :
oEther oxyde primaire ou sec. + HX alcool (+substitué) + RX (- substitué)→
oEther oxyde tertiaire + HX alcool (- substitué) + RX (+ substitué)→
Synthèse des Phénols :
oBenzène + HSO3+ cycle-HSO3 + Base Phénol.→ →
Réaction des Phénols :
oBromation
oSulfonation…
oPhénol + RX éther oxyde (avec le cycle)→
oOxydation des phénols Quinone→
Epoxydes :
oAlcène + Acide peroxycarbolique (cycle-C=O-OOH) Epoxyde (sur l’alcène)→
oEpoxyde + H3O+ Diol (Trans)→
Thiol et sulfure :
oRX + NaSH Thiol + Na X→
oRSH + Na RS- Na + H2→
RS- + RX Sulfure + X Na→
Chapitre 9 :

Les Aldéhyde et les cétones sont des composés particuliers, ils font parti de la famille des composés
carbonylés. Leur réactivité est particulière puisque les substituants H ou R sont de mauvais groupes
partant, il ne peuvent stabilisés une charge -, contrairement au reste des composés carbonylé. L’oxygène
de la boule liaison avec le carbone est hybridé sp2. (Les Aldéhydes sont tout de même plus réactif.)
3 Aldéhydes a connaître : Le formaldéhyde ou Formol ou méthanal (HCHO), l’Acétaldéhyde (CH3CHO), le
Benzaldéhyde (cycle-CHO).
3 Cétones a connaître : L’Acétone (CH3COCH3), l’Acétophénone (cycle-COCH3), Benzophénone (cycle-
CO-cycle).
Les réactions :
Synthèse des aldéhydes et cétones :
oDéjà vu : Oxydation des alcools (primaires ou secondaires), hydratation des alcynes,
acylation de Friedel-Craft.
Réaction :
oOxydation des aldéhydes en présence du réactif de Tollens (NH3Ag+) acide carbo→
Impossible pour les cétones
oRéaction général d’addition nucléophile : Attaque du nucléo. , carbocation avec stabilisation
de la charge – sur l’oxygène, et attaque d’un hydrogène sur l’oxygène.
oAddition basique de l’eau Diol →
(Mécanismes différents)
oAddition acide de l’eau Diol→
oAddition d’un alcool Acétal →
(Mécanisme important -> passage par un Hémiacétal)
Cette réaction permet de protéger le groupe aldéhyde et les cétones, la réaction étant
totalement réversible.
o Addition d’une amine Imine →
(!!! Cela provoque le départ du groupe =O, remplacer par =N-R)
oAddition d’un Hydroxylamine (NH2OH) Oxyme ( =NOH)→
oAddition d’un réactif de Grignard Alcool→
On peut formé un réactif de Grignard avec R, aldéhyde, cétone, amide, NO2, CN, SO2R→
On ne peut pas formé de réactifs avec OH, NH, SH, COOH.→
Chapitre 10 :
Parmi le groupe des acides carboxyliques on distingue, les halogénures d’acyles, les anhydrides d’acide,
les esters, les amides et les nitriles.
Un caractère particulier des acides carboxyliques est qu’ils sont acides, bien plus acide que les alcools par
exemple. Leur acidité dépend tout de même du substituant à savoir si il est plutôt électro-attracteur ou pas,
donc sa capacité a stabiliser la charge -.
Une autre de leur particularité et que le groupe ROR, NH2, OH…accroché au carbone stabilise les charges –
et son ainsi de bon groupe partants.
Ils ont des points d’ébullition élevé car ils peuvent faire des pont hydrogènes entre eux.
Réaction générale de substitution nucléophile des composés carbonylés :
Attaque du nucléophile, stabilisation de la charge – par O, expulsion du groupe carbonyle.
Les Réactions :
Synthèse des acides carboxyliques :
oDéjà vu : Alcène en présence de KMnO4, cycleR en présence de KMnO4, alcool primaire
oxyde par CrO3, aldéhyde oxydé par AgNH3.
oNa+ CN- + RX Nitriles en présence d’acide ou de base acide carbo.→ → →
oRéactif de Grignard + CO2 acide carbo.→
Réactions des acides carboxyliques :
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%