Métabolisme des acides aminés et de l`azote

UE 3 – Biochimie clinique, Nutrition, Métabolisme
Gonthier
!
Date : 21/09/2015 Plage horaire : 16h00-18h00
Promo : DCEM1 Enseignant : Dr M-P. Gonthier
!
Ronéistes :
ESTERMANN Laurine
LESQUERRE-CAUDEBEZ Alizée
!
!
!
Métabolisme des acides aminés et de l’azote
!
I.Introduction
1.Généralités
2.Structure des acides aminés
3.classifications des acides aminés
4.Besoins en AA indispensables chez l'Homme (en mg/kg/j)
!
II.Digestion des protéines alimentaires
1.Digestion intraluminale
2.Absorption entérocytaire des AA.
3.Maladies congénitales dues aux systèmes de transport
4.Métabolisme entérocytaire
5.Les différents types de catabolisme des protéines
6.La dégradation lysosomiale des protéines
7.Dégradation des protéines cellulaires par le système ubiquitine-
protéasome (dans le cytoplasme)
8.Transport des aa (via le sang)
9.Métabolisme hépatique
!
III.Catabolisme des acides aminés
1.Catabolisme du squelette carboné : plusieurs devenirs en fonction des
situations physiologiques
2.Catabolisme de la fonction amine NH₂
3.Devenir de la chaîne carbonée de l’acide aminé ?
4.Bilan du devenir catabolique des acides aminés
5.Enzymes clés du métabolisme des acides aminés
6.Echange inter-organes en situation de jeûne (couple alanine/glucose)
! sur ! 1 33

!
IV.Biosynthèse des acides aminés et dérivés
1.Biosynthèse des acides aminés non essentiels
!
!
2. Biosynthèse des dérivés d’acides aminés (fin ronéo)
3. Biosynthèse de la créatinine à partir de glycine et arginine
4. Biosynthèse du glutathion par condensation du glutamate, cystéine et glycine
5. Conversion des acides aminés en amines biologiques par décarboxylation – synthèse de NO
!
V. Pathologies associées au métabolisme des acides aminés et de l’azote
VI.Exploration du métabolisme azoté!
!
!!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
! sur ! 2 33

!
Dans un premier temps, nous reverrons quelques généralités sur les AA, leur structure… Puis nous
verrons la notion de digestion des protéines alimentaires aux niveaux du tube digestif et intra-cellulaire
(système ubiquitine-protéasome). Ensuite nous aborderons le catabolisme des AA, qui générera de
l’énergie puis leur biosynthèse : il n'existe pas de stock d’AA libres ! Les AA qui arrivent en excès dans
l'organisme sont en effet stockés sous forme de protéines :
−catabolisme des AA en situation de besoin énergétique,
−synthèse de protéines en situation de suffisance énergétique.
On verra ensuite les différentes pathologies associées à un défaut du métabolisme des AA et de l’azote.
Enfin, on donnera quelques pistes sur les moyens d'explorer au laboratoire ce métabolisme (à partir de
marqueurs cliniques), ce qui nous permettra de diagnostiquer certaines pathologies.
!
!
I.Introduction
!
1.Généralités
!
Les acides aminés dans la cellule peuvent être :
!
- source d’énergie, car ils possèdent un pouvoir énergétique intrinsèque et peuvent donc servir de
substrat énergétique/source d'énergie pour les cellules. Consommer 1g de protéines permet d'apporter
4kCal d’énergie. Les AA issus des protéines alimentaires permettent de satisfaire 15 à 20% de nos
besoins énergétiques (glucides: 50%; lipides: 30%).
!
- précurseurs d'hormones et médiateurs cellulaires : rôle dans la régulation des grandes
fonctions physiologiques. Ils sont à l'origine de nombreux messagers intra ou inter-cellulaires (dont les
neuromédiateurs). Ex: dopamine, glutathion…
!
- ds éléments constitutifs de protéines que notre organisme va devoir synthétiser dans le but
d’assumer toutes nos fonctions physiologiques.
!
L’origine des acides aminés est double :
- Les AA peuvent provenir du catabolisme des protéines alimentaires (catabolisme au niveau de
l'estomac puis du duodénum et de l'intestin).
!
Ronéo 2014
La plupart des protéines vont être dégradées : 95% des AA passeront la barrière intestinale (sous forme
libre) et arriveront dans la veine porte et le foie. 2% des protéines alimentaires pourront traverser la paroi
intestinale : il s'agit des anticorps provenant du lait maternel. Puis les tissus cibles reconstitueront les
protéines. « Donc ce n'est pas la protéine du steak de bœuf qui va se stocker directement dans les muscles,
bien évidemment... »
!
- Il peut aussi y avoir une synthèse de novo : les acides aminés libres pourront être produits lors
du catabolisme de protéines endogènes et lors de l'interconversion des substrats énergétiques dans le
schéma complexe du métabolisme énergétique.
!
Exemple : on peut obtenir de l'alanine à partir de glucose : glucose → pyruvate → alanine.
!
! sur ! 3 33

Le métabolisme résulte d’un équilibre dynamique entre le pool d'acides aminés et le pool de protéines
dans l’organisme.
!
AA qui arrivent dans l'organisme en excès :
−une partie sera stockée sous forme de protéines,
−l'autre partie directement catabolisée pour donner de l’énergie car pas de stock d’AA sous forme
libre.
!
2.Structure des acides aminés
!
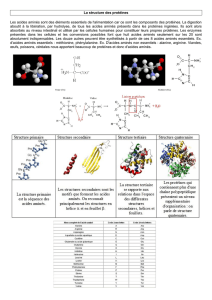
Les acides aminés sont des acides carboxyliques α-aminés. Ce sont des molécules organiques, avec un
squelette carboné, qui se caractérise par un carbone (central) α tétravalent dit asymétrique ayant des
liaisons avec :
−une chaîne latérale R : spécifique de chaque AA,
−une fonction COOH (= partie carboxylique) d’où le nom « d’acide »,
−une fonction NH2 (= fonction amine, qui contient l'atome d’azote) d’où le nom d’ « aminé »,
−un atome d'hydrogène (pour compléter la tétravalence de l'atome de carbone).
!
20 AA sont dits protéinogènes, servent donc à la fabrication des protéines. 2 de plus (l’ornithine et la
citrulline, dérivants tous les deux de l’arginine) ne sont pas impliqués dans la fabrication des protéines
sous forme de liaisons peptidiques, mais dans le cycle de l’urée (dans l’uréogenèse au niveau du foie).
!
Il existe 20 AA naturels ayant tous la structure citée ci-dessus, SAUF la glycine. La glycine est un AA à
part car sa chaîne latérale R est un H. Elle n’est donc ni polaire, ni apolaire et n'a pas de carbone
asymétrique.
!
•La fonction carboxylique correspondant au carbone α et au reste de la chaîne carbonée va
donner de l'énergie au moment du catabolisme des AA.
!
•La fonction NH₂ correspond au groupement amine et l'atome d'azote va nécessiter un
traitement particulier, car ce NH₂ sera susceptible de donner du NH₃ (ammoniac) qu’il faudra
immédiatement éliminer par le cycle de l'uréogenèse (cycle de l'urée).
!
•Le radical R permet à chaque AA d’avoir des propriétés physico-chimiques propres en termes
de polarité, de charge électrique et de groupe réactif acide ou basique (groupement fonctionnel).
!
L’AA peut s’ioniser : NH3+ - CH - COO-.
!
#
!
! sur ! 4 33

Concernant la structure des 20AA protéinogènes
•AA apolaires : insolubles dans l’eau, hydrophobes. Retrouvés en général au coeur des protéines.
•AA polaires ou hydrophiles : retrouvés à la surface pour interagir avec le solvant aqueux. Parmi ces
AA polaires, certains sont neutres, d’autres chargés positivement dits basiques, d’autres chargés
négativement dits acides.
!
!
3.Classifications des acides aminés
!
Il existe plusieurs sortes de classifications.
!
A.Selon leur structure
!
D’après cette classification, 20 acides aminés naturels participent à la structuration / fabrication des
protéines. Ils sont classés selon leur degré de polarité (donc selon la nature de la chaîne latérale R).
!
#
!
Ronéo 2014
Ils sont d'origine alimentaires. Les AA peuvent être polaires (chargés ou non chargés), ou apolaires : on a
les 2 AA soufrés, les AA aromatiques et les AA aliphatiques.
!
! sur ! 5 33
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
1
/
33
100%