echos_pharmaceutiques17_n°22 octobre 2014

N
N
NO
O
OU
U
UV
V
VE
E
EA
A
AU
U
UT
T
TÉ
É
ÉS
S
S
T
T
TH
H
HÉ
É
ÉR
R
RA
A
AP
P
PE
E
EU
U
UT
T
TI
I
IQ
Q
QU
U
UE
E
ES
S
S
Lixisenatide (LYXUMIA
®
) et diabète de type 2
Après Victoza
®
(liraglutide) et Byetta
®
(exénatide), Lyxumia
®
(lixisénatide) est le 3
ème
analogue
des incrétines à obtenir une AMM dans l’Union Européenne. Son action agoniste des récepteurs du
GLP-1 potentialise de façon glucose-dépendante la sécrétion d'insuline par les cellules bêta du
pancréas et inhibe la sécrétion de glucagon dans les cellules alpha pancréatiques. En cas
d'hypoglycémie, le mécanisme de secours que constitue la sécrétion de glucagon est donc préservé.
Ce médicament est indiqué dans le traitement du diabète de type 2, en association avec les
antidiabétiques oraux et/ou une insuline basale, lorsque ceux-ci n’ont pas permis d’atteindre un
contrôle glycémique adéquat.
Il se présente sous forme de stylo pour injection sous-cutanée et s’emploie à la posologie de 10µg
une fois par jour pendant 14 jours puis en entretien à 20 µg une fois par jour.
Cependant, le lixisénatide semble partager les nombreux effets indésirables des analogues des
incrétines. Il s’agit principalement de nausées, vomissements, diarrhées, insuffisances rénales,
troubles thyroïdiens et quelques cas de pancréatites.
Le laboratoire sollicite une ASMR V en présentant le lixisénatide comme une « alternative
supplémentaire aux agonistes des récepteurs du GLP-1 commercialisés. Par rapport à Byetta,
Lyxumia
®
, dont la non infériorité a été démontrée sur la diminution de l’HbA1c, présente un profil
de tolérance amélioré en termes de réduction des événements gastro-intestinaux et de réduction des
hypoglycémies symptomatiques. Son administration en une injection par jour (au lieu de deux
injections pour Byetta) en fait une alternative supplémentaire utile ».
Retiré du marché allemand au motif d’un prix de remboursement insuffisant, des nouvelles études
sont en cours pour démontrer son efficacité sur les critères cliniques de mortalité et de morbidité
cardiovasculaire.
Sources : Commission de la Transparence du 8/01/2014 ; Prescrire Mai 2014
Rédacteur : Marion Bessone - Interne-HIA Sainte-Anne-Toulon
Dolutégravir (TIVICAY®) et VIH de type 1
Le traitement de première intention des infections par le virus de l’Immunodéficience Humaine de
type 1 (HIV-1) est relativement consensuel dans son principe. Il repose sur une association de trois
antirétroviraux issus de 2 classes thérapeutiques différentes. Les inhibiteurs de l’intégrase, classe
destinée le plus souvent aux patients en situation d’échec thérapeutique, étaient jusqu'à récemment,
disponibles en France au nombre de deux (raltégravir ISENTRESS® et elvitégravir VITEKTA®).
Depuis le 21 janvier 2014, le dolutégravir, nouvel inhibiteur de l’intégrase élaboré par le
laboratoire ViiV Healthcare, a obtenu l’AMM par l’EMA en combinaison avec d’autres agents
antirétroviraux pour le traitement des adultes et adolescents de plus de 12 ans atteints par le HIV-1.
En France, cette molécule est disponible depuis le 5 mai 2014 à l’hôpital en relais d’ATU
nominatives. Chez les patients dont le virus ne possède pas de mutation de résistance aux
inhibiteurs de l’intégrase, le dolutégravir a montré une efficacité immuno-virologique non
inférieure à celle du raltégravir avec une barrière génétique au développement de résistance plus
élevée. Chez les patients en impasse thérapeutique et dont le virus est sensible au dolutégravir, ce
dernier apporte un progrès thérapeutique modéré en terme d’efficacité immuno-virologique. Son
mécanisme d’action repose sur l’inhibition de l’intégrase, enzyme nécessaire pour incorporer
l’ADN viral dans le matériel génétique de la cellule infectée. Ce comprimé jaune dosé à 50 mg,
présente un double avantage pour le patient : facilité d’administration (une prise quotidienne per os,
de préférence à heure fixe) et peu d’effet indésirable (rash peu fréquent).
Notons que cette molécule se retrouve également au sein d’une nouvelle spécialité TRIUMEQ®
(abacavir 600 mg/ dolutegravir 50 mg/ lamivudine 300 mg), qui vient d’obtenir l’AMM de l’EMA
le 10 septembre 2014. Bientôt sur le marché français, cette spécialité présentera l’avantage de
regrouper la trithérapie antirétrovirale au sein d’un seul comprimé à prendre quotidiennement.
Références : Synthèse d’avis de la commission de la transparence de l’HAS- Mai 2014 ; Prescrire Juin 2014 ; Rapport sur
l’épidémie mondiale de sida 2013 ONUSIDA
Rédacteur : Alice VIALLET – ERSA Marolles
N
N
NO
O
OU
U
UV
V
VE
E
EA
A
AU
U
UT
T
TÉ
É
ÉS
S
S
L
Li
ix
xi
is
se
en
na
at
ti
id
de
e
(LYXUMIA
®
)
e
et
t
d
di
ia
ab
bè
èt
te
e
d
de
e
t
ty
yp
pe
e
2
2
D
Do
ol
lu
ut
té
ég
gr
ra
av
vi
ir
r
(TIVICAY
®
) e
et
t
V
VI
IH
H
d
de
e
t
ty
yp
pe
e
1
1
V
V
VI
I
IG
G
GI
I
IL
L
LA
A
AN
N
NC
C
CE
E
ES
S
S
G
Ga
ab
ba
ap
pe
en
nt
ti
in
ne
e
e
et
t
p
pr
ré
ég
ga
ab
ba
al
li
in
ne
e
INFORMATIONS
N
Ne
ef
fo
op
pa
am
m
e
et
t
u
ut
ti
il
li
is
sa
at
ti
io
on
n
i
in
nt
te
en
ns
si
iv
ve
e
L
La
a
b
bi
il
lh
ha
ar
rz
zi
io
os
se
e
e
en
n
C
Co
or
rs
se
e
e
et
t
l
le
es
s
p
pr
ro
ob
bl
lè
èm
me
es
s
d
d’
’a
ap
pp
pr
ro
ov
vi
is
si
io
on
nn
ne
em
me
en
nt
t
O
O
C
C
T
T
O
O
B
B
R
R
E
E
2
2
0
0
1
1
4
4
N°
22
L
LE
ES
S
É
ÉC
CH
HO
OS
S
P
PH
HA
AR
RM
MA
AC
CE
EU
UT
TI
IQ
QU
UE
ES
S
D
DE
ES
S
H
HI
IA
A
Comité de rédaction :
- PhC HOFMANN : HIA LEGOUEST
- Ph SPADONI : HIA SAINTE-ANNE

V
V
VI
I
IG
G
GI
I
IL
L
LA
A
AN
N
NC
C
CE
E
ES
S
S
Gabapentine et prégabaline : la notification spontanée au service de la pharmacovigilance
La gabapentine (Neurontin
®
) et la prégabaline (Lyrica
®
) sont des molécules analogues de l’acide gamma-aminobutyrique La gabapentine est
indiquée dans les crises d’épilepsie partielle et les douleurs neuropathiques sévères alors que la prégabaline peut être également utilisée dans les
troubles anxieux généralisés. Une étude parue en 2013 a analysé les déclarations de pharmacovigilance parues entre le 1
er
janvier 1995 et le 31
décembre 2009 ce qui a permis de mettre en évidence des effets indésirables.
Au total 2415 effets indésirables ont été rapportés : 725 patients pour la gabapentine et 608 patients pour la prégabaline.
Les principaux effets observés sont les troubles neuropsychiques : somnolence, sensations vertigineuses, agitations, comportements agressifs
mais aussi confusions et hallucinations. Viennent ensuite les troubles hépatiques ainsi que les troubles sanguins.
En effet 12% et 5% des cas notifiés concernent respectivement une atteinte hépatique sous gabapentine et sous prégabaline avec 3 décès suite à
une hépatite fulminante.
7% et 8% des cas notifiés concernent respectivement une atteinte de la lignée sanguine sous prégabaline avec prédominance des leucopénies,
agranulocytose, thrombopénie et sous gabapentine.
Des effets cutanés, immunoallergiques et cardiovasculaires ont également été observés.
On note également l’apparition d’effets indésirables chez des enfants exposés in utero mais ces notifications ne permettent pas de tirer une
conclusion car seules les expositions associées à une malformation sont notifiées.
Enfin concernant la pharmacodépendance, même si le nombre de cas d’abus et de pharmacodépendance rapportés en France est faible, il est
fréquent dans les pays du Nord de l’Europe ce qui justifie l’ouverture d’un suivi prospectif sous forme d’un suivi national d’addictovigilance
avec mise en place d’échanges réguliers entre le laboratoire et le CEIP
1
rapporteur.
Sources :
1
Comité technique des Centres d’Evaluation et d’Information de la Pharmacodépendance-Séance du 11 juillet 2013
;
Prescrire Juillet 2014
Rédacteur : Ph SPADONI Sophie -HIA Sainte-Anne-Toulon
I
IN
NF
FO
OR
RM
MA
AT
TI
IO
ON
NS
S
P
PH
HA
AR
RM
MA
AC
CE
EU
UT
TI
IQ
QU
UE
ES
S
Utilisation intensive du Néfopam (Acupan®) : la fin justifie – t – elle les moyens ?
Le Néfopam est un antalgique autorisé en France depuis 1980 par voie injectable et orale (d’ailleurs utilisé hors AMM). Très utilisé avec plus de
2,5 millions de boites remboursées en 2013, il présente un mode d’action original par rapport aux autres antalgiques, mêlant des effets
atropiniques et dopaminergiques. Toutefois son efficacité reste mal établie, par des essais cliniques anciens et de qualité méthodologique
incertaine. Elle semble être au mieux modeste, probablement pas supérieure à celle des AINS. Il est néanmoins vraisemblable que certains
patients se sentent globalement mieux soulagés par le Néfopam en raison de ses propriétés psychostimulantes. Mais ces mêmes propriétés
exposent à un risque d’abus avec développement de dépendances même chez des patients sans antécédent. En revanche ses effets indésirables,
même aux doses recommandées, sont bien connus : effets anticholinergiques et sensations vertigineuses, troubles neuropsychiques et réactions
graves d’hypersensibilité. En outre, il est métabolisé par le foie et éliminé par voie rénale, ce qui induit une variabilité prévisible d’efficacité et
de tolérance en cas d’interaction avec les inducteurs enzymatiques et chez l’insuffisant rénal. En bref plus de trente ans après sa
commercialisation, faute d’évaluation suffisante, la balance bénéfice-risque du Néfopam est mal cernée avec des effets indésirables et un
potentiel addictif mieux établis que son efficacité. Cela conduit à recommander son emploi de façon limitée, uniquement sur de courtes durées et
chez des patients informés des risques d’intolérance et des incertitudes relatives à son efficacité…
Sources :Prescrire Septembre 2014
Rédacteur : Ph LEFEUVRE Leslie -HIA Legouest-Metz
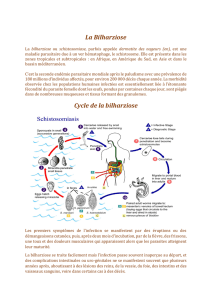
La bilharziose en Corse et les problèmes d'approvisionnement
La bilharziose (ou schistosomiase) est une maladie provoquée par une famille de vers parasites, les schistosomes. Ces derniers sont présents
dans certaines eaux douces des zones tropicales et subtropicales, notamment en Afrique. La bilharziose peut prendre deux formes principales :
intestinale et urogénitale. Il s’agit de la maladie parasitaire la plus répandue après le paludisme, elle tuerait ainsi près de 300 000 personnes par
an. Il existe pourtant un traitement efficace et bien toléré : le praziquantel (Biltricide
®
, comprimés de 600mg). La posologie varie en fonction du
parasite ; à titre d’exemple, elle est de 40 mg/kg en une prise unique pour Schistosoma haematobium.
Deux situations inhabituelles se sont cumulées en 2014. Dix-sept cas de bilharziose urogénitale ont été diagnostiqués en Corse suite à une
baignade dans la rivière Cavu, ce qui a donné lieu à des recommandations de l’HCSP et de l’ANSES visant à éviter la propagation d’une
épidémie. C’est dans ce contexte qu’est survenue au mois de juillet une perturbation de l’approvisionnement de Biltricide
®
en France, suite à un
problème de fabrication. Afin de conserver la capacité de traitement d’une épidémie de bilharziose en Corse, l’ANSM a recommandé aux
médecins prescripteurs de réserver la prescription de praziquantel aux indications de l’AMM (bilharzioses et distomatoses) pour les cas où la
pathologie est confirmée biologiquement. D’autre part, la spécialité de praziquantel destinée au marché ukrainien a été mise à disposition des
PUI, avec autorisation de la rétrocéder aux patients ambulatoires. La rupture de stock du Biltricide® a pris fin à partir du 18/08/2014.
Sources : http://www.sante.gouv.fr/bilharziose.html ; http://www.ansm.sante.fr
Rédacteur : Vanessa Polly-HIA Percy
P
OUR PLUS D
’
INFOS OU POUR SUGGERER
DE NOUVELLES IDEES
,
N’
HESITEZ PAS A NOUS CONTACTER
!
M
ME
HOFMANN
C
HRISTELLE
HIA
L
EGOUEST
03
87
56
22
81
1
/
2
100%