Origine cardiaque des malaises

Origine cardiaque
des malaises
Vincent Lucet
Unité de rythmologie pédiatrique, Château des Côtes, 78350 Les Loges-en-Josas
La survenue d’un malaise inopiné chez un nourrisson, éventuellement premier-
né, est la source d’une angoisse considérable chez de jeunes parents. Un exa-
men clinique normal au décours est apriorirassurant mais ne permet pas
d’éliminer formellement une pathologie cardiaque, qu’il s’agisse d’une cardio-
pathie muette (non soufflante) ou d’un trouble du rythme paroxystique. Au
moindre doute, la réalisation d’une échographie cardiaque, d’un électrocardio-
gramme et, éventuellement, d’un enregistrement de Holter, doit être proposé.
À cet âge, les principaux « pièges diagnostiques » sont l’hypertension artérielle
pulmonaire (primitive ou secondaire à une cardiopathie congénitale méconnue),
l’existence d’une coronaire gauche anormale ou d’une myocardiopathie. On
doit aussi rechercher, par principe, un trouble du rythme paroxystique méconnu
(tachycardie jonctionnelle réciproque, syndrome de QT long ou de Brugada,
bradycardie paroxystique par bloc auriculo-ventriculaire ou hyperréactivité
vagale).
Mots clés : malaise, hypertonie vagale, cardiopathie congénitale, arythmie, mort subite du
nourrisson
Les malaises constituent une cause
fréquente de consultation pédia-
trique. Parfois, leur gravité apparente
justifie l’appel des secours d’urgence
(pompiers –Samu) et l’admission
directe en milieu pédiatrique spécia-
lisé [1]. Il est fréquent cependant que
le pédiatre de ville soit consulté à pro-
pos d’un incident qui a inquiété la
famille. L’interrogatoire et l’examen
clinique permettent alors d’effectuer
un premier tri : on prescrit au moindre
doute un bilan complémentaire sélec-
tif, parmi lequel se discute l’utilité
d’explorations cardiaques (ECG, Hol-
ter, réflexe oculocardiaque et écho-
graphie). Les cardiopathies « non
soufflantes », les troubles du rythme
supra ou ventriculaires et l’hyperréac-
tivité vagale constituent les 3 causes
principales des malaises d’origine car-
diaque à rechercher chez le nourris-
son [2].
Cardiopathies
non soufflantes
Si l’existence d’un gros souffle,
d’une cyanose, de signes évidents de
défaillance cardiaque conduisent
directement à une prise en charge spé-
cialisée, l’absence de souffle chez un
nourrisson eupnéique ne permet pas
d’éliminer formellement une cardio-
pathie silencieuse. Surtout si l’enfant
pousse mal, si les biberons sont labo-
rieux, si le malaise a eu un caractère
inquiétant, il faut penser, par principe,
à la possibilité d’une cardiopathie. À
titre d’exemple, nous évoquerons ici
quelques diagnostics particulièrement
difficiles dont l’expérience nous a
appris à nous méfier, après plusieurs
dizaines d’années de travail dans un
hôpital de jour spécialisé dans l’explo-
ration des malaises cardiaques de
l’enfant.
m
t
p
Tirés à part : V. Lucet
doi: 10.1684/mtp.2009.0219
mt pédiatrie, vol. 12, n° 2, mars-avril 2009
Dossier
98
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

Coronaire gauche anormale
Il s’agit du type même de la cardiopathie non souf-
flante qui peut se révéler, après un intervalle libre de quel-
ques semaines, par un malaise brutal avec pâleur et ady-
namie chez un nourrisson paraissant jusque-là bien
portant [3]. En effet, en période néonatale, la pression pul-
monaire reste suffisamment élevée pour permettre une
perfusion coronaire adaptée. Dès que les résistances pul-
monaires baissent, la perfusion coronaire gauche diminue
et un infarctus antérolatéral peut survenir. Cliniquement,
cet infarctus brutal s’accompagne souvent d’un malaise
précédé de cris anormaux et accompagnés de pâleur.
L’examen clinique au décours peut être anormal (tachy-
pnée, tachycardie, bruit de galop, gros foie), mais est sou-
vent sensiblement normal. L’électrocardiogramme montre
typiquement des ondes Q de nécrose en D1, AVL et de
V4 à V6 avec courant de lésion et signe d’ischémie.
Cependant, l’ECG à cet âge est souvent sans particularité
et sa normalité n’élimine pas le diagnostic. En fait, c’est
l’échographie qui permet d’évoquer le diagnostic, mon-
trant un tableau de myocardiopathie dilatée, akinétique,
avec une petite insuffisance mitrale et des piliers mitraux
hyperéchogènes. La coronaire gauche est mal vue alors
que la coronaire droite peut être dilatée du fait du déve-
loppement d’un réseau collatéral. Les enzymes musculai-
res et la troponime sont élevés. L’enfant doit être dirigé
vers un service de réanimation chirurgicale pour une
prise en charge préopératoire active et la confirmation,
si nécessaire, du diagnostic par aortographie, ou mieux
maintenant : scanner multibarette ou IRM.
Hypertension artérielle pulmonaire
L’hypertension artérielle pulmonaire, qu’elle soit pri-
mitive ou secondaire à une cardiopathie, est source
potentielle de malaises avec cyanose, parfois pâleur et
perte de connaissance pouvant survenir à l’effort (bibe-
ron) ou aux cris (on évoque alors souvent le diagnostic
de spasmes du sanglot), ou, de façon parfois plus évoca-
trice, la nuit, au cours du sommeil. Malgré le caractère
fréquemment répétitif de ces malaises, le diagnostic
d’hypertension artérielle pulmonaire est souvent porté
avec retard [3]. On évoque, classiquement en premier
lieu, un malaise lié à un reflux gastro-œsophagien (surtout
si les incidents surviennent à l’occasion du biberon), des
spasmes du sanglot ou une hyperactivité vagale. De fait,
l’examen clinique est souvent pauvre : il s’agit aussi d’une
affection cardiaque « non soufflante », et l’éclat du 2
e
bruit pulmonaire ainsi que le petit souffle diastolique de
haute fréquence (témoin de l’insuffisance pulmonaire)
sont très inconstants chez le nourrisson. De plus, pendant
plusieurs mois au moins, les signes de décompensation
droite peuvent être absents, et on ne note ni tachycardie
importante, ni bruit de galop, ni gros foie, ni dyspnée de
repos : c’est-à-dire qu’il peut s’agir d’un diagnostic parfois
difficile, à évoquer par principe chez le nourrisson, si les
circonstances de malaises (évoqués plus haut) sont retrou-
vées et à la moindre petite anomalie de l’examen clinique
ou devant un retard staturo-pondéral avec cassure de la
courbe de croissance. L’hypertension artérielle pulmo-
naire primitive est relativement exceptionnelle mais peut
alors être familiale. Le plus souvent, l’HTAP est secon-
daire à une cardiopathie. Les shunts gauche-droit avec
égalité de pression entre la cavité gauche et droite repré-
sentent la plus fréquente des causes de l’HTAP : le canal
artério-ventriculaire complet, les très gros canaux arté-
riaux, les larges CIV, ou les retours veineux pulmonaires
anormaux totaux et dans une moindre mesure, les très
larges CIA sont les cardiopathies le plus souvent retrou-
vées à l’échographie dans ce contexte. Il peut aussi
s’agir d’un obstacle post-capillaire gênant l’écoulement
du sang veineux pulmonaire et responsable d’une HTAP
post-capillaire. Une telle situation peut se rencontrer en
cas de sténose congénitale isolée des veines pulmonaires,
de cœur triatrial, de sténose mitrale ou d’anomalie de la
compliance du ventricule gauche. Quoi qu’il en soit, c’est
l’échographie-Doppler qui fait le diagnostic, montrant
souvent un ventricule droit dilaté ou hypertrophié, avec
une courbure septale anormale en systole (plate ou
convexe de droite à gauche) témoignant d’une tendance
àl’égalisation de la pression entre les ventricules gauche
et droit (figure 1).L’enregistrement d’une fuite tricuspide
véloce (4 à 5 mm/sec) permet d’objectiver l’augmentation
de la pression systolique pulmonaire. De même, une fuite
pulmonaire véloce en protodiastole permet d’objectiver
un gradient diastolique élevé entre le ventricule droit et
l’artère pulmonaire. L’analyse échographique des lésions
associées permet de confirmer l’HTAP et d’en trouver la
cause. C’est-à-dire l’importance de la réalisation d’une
échographie cardiaque au moindre doute, devant un
malaise un peu insolite, ou récidivant sans cause retrou-
vée, chez un nourrisson (tableau 1).
Troubles du rythme paroxystique
Rythmes réciproques paroxystiques
et syndromes de Wolff. Parkinson White
Les tachycardies jonctionnelles réciproques paroxysti-
ques (TJR) représentent 70 à 80 % des tachycardies supra-
ventriculaires du nourrisson. Elles sont plus fréquentes
chez les garçons que chez les filles, et une cardiopathie
(Ebstein, double discordance, myocardiopathie, etc.) est
présente dans 10 à 15 % des cas, justifiant la pratique
systématique d’une échographie lors de la découverte
du trouble du rythme. Le mode de révélation le plus fré-
quent est l’insuffisance cardiaque (60 % des cas avant
mt pédiatrie, vol. 12, n° 2, mars-avril 2009 99
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

l’âge de 3 mois). Mais le trouble du rythme peut être
découvert à l’occasion d’un malaise brutal avec pâleur,
pleurs non justifiés, refus du biberon, troubles digestifs.
La gravité habituelle de la présentation initiale s’oppose
au bon pronostic final chez le nourrisson. Qu’il y ait ou
non des signes patents de préexcitation (PEV), les TJR du
nourrisson sont en rapport, dans 60 à 80 % des cas, avec
une voie accessoire de préexcitation [4]. En cours
d’accès, la fréquence ventriculaire est rapide (250 à
320 bats/mn) avec, le plus souvent, une onde P’rétro-
grade située à distance du QRS (RP’≥80 ms). La prise
en charge initiale comporte, si nécessaire, le rétablisse-
ment des grandes fonctions vitales en service de réanima-
tion, et/ou la réduction en urgence du trouble du rythme
par manœuvres vagales (compression oculaire, vessie de
glace sur le visage, injection rapide d’adénosine intravei-
neuse). L’arrêt brutal de la tachycardie n’empêche pas
une récidive précoce ou plus tardive de l’arythmie et jus-
tifie la mise en route d’un traitement préventif : la digo-
xine doit être utilisée avec prudence dans ce contexte
(10 gammas/kg/jour en 3 prises, sans doses de charge)
(tableau 2). Associé à un traitement bêtabloquant (acébu-
tolol pédiatrique ou Bisoprolol
®
), la digoxine peut suffire
pour éviter les récidives. Au moindre doute, on utilisera
l’amiodarone. Le traitement d’entretien est poursuivi
jusqu’àl’âge de 1 an en moyenne. L’apparition d’une dys-
thyroïdie peut conduire à remplacer l’amiodarone par un
antiarythmique de classe I (flécaïnide ou propafénone).
Le pronostic des TJR du nourrisson est bon avec 70 à
80 % de guérison, sans récidive au-delà de 1 an. Les quel-
ques cas de mort subite [5] semblent en rapport avec des
troubles du rythme négligés ou avec un surdosage digita-
lique (figure 2). Un traitement du fond (bêtabloquant,
antiarythmique de classe I) peut être prescrit pendant
quelques années chez le nourrisson ou le petit enfant
symptomatique, avant d’envisager une exploration par
Figure 1. Nourrisson de 18 mois : équivalent répétés et graves de spasmes du sanglot (cyanose) et survenue de malaises nocturnes :
l’échographie objective une nette dilatation du ventricule droit (R.V), refoulant le ventricule gauche (LV) : il s’agit d’une hypertension arté-
rielle pulmonaire liée à une sténose des veines pulmonaires associée à un cœur tri-atrial.
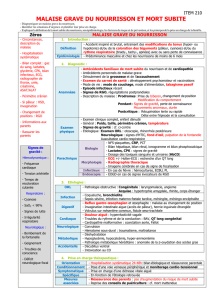
Tableau 1. Principale cardiopathie pouvant se reveler
par un malaise chez un nourrisson
Hypertension artérielle pulmonaire :
•primitive (rare)
•secondaire à un shunt gauche-droit (canal atrio-ventriculaire,
communication inter-ventriculaire, retour veineux pulmonaire anormal,
canal artériel, troncus …)
•secondaire à un obstacle post-capillaire (sténose des veines pulmonaires,
cœur triatrial, sténose mitrale…)
Cardiopathie cyanogène :
•Tétralogie de Fallot
•Atrésie pulmonaire
•Ventricule unique
Coronaire gauche anormale (++)
Myocardiopathie
Tumeur du cœur (rare)
mt pédiatrie, vol. 12, n° 2, mars-avril 2009
Origine cardiaque des malaises
100
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

voie œsophagienne et l’ablation de la voie accessoire par
radiofréquence si la période réfractaire antérograde est
courte (≤220 ms). Les réentrées intranodales sont excep-
tionnelles avant l’âge de 2 ans [6] et leur fréquence ne
dépasse pas 20 à 30 % de l’ensemble des TJR de l’enfant.
Il s’agit aussi d’une tachycardie supraventriculaire paro-
xystique, rapide (± 300/mm), sans onde P’individuali-
sable en cours d’accès. Sa prise en charge médicale
dépend de la gêne fonctionnelle qu’elle procure et peut
motiver une ablation par radiofréquence à l’adolescence.
Des formes familiales de Wolff-Parkinson-White (WPW) à
transmission autosomique dominante ont été décrites et
un gène récemment identifié [7]. L’incidence d’un WPW
familial parmi les patients qui ont à l’ECG une voie acces-
soire patente est de 34/1 000 avec une prévalence de
préexcitation parmi les membres de la famille du 1
er
degré de ces patients qui est de 5,5/1 000 personnes,
significativement plus importante que la prévalence du
WPW [17] dans la population générale (1 à 3/1 000). Glo-
balement, les TJR du nourrisson ont un bon pronostic final
en l’absence de cardiopathie sous-jacente. Une bonne
prise en charge médicale permet en principe d’éviter la
survenue d’accident grave ou d’une mort subite [8].
Les indications d’ablation doivent être prises à temps
chez les grands enfants mais comportent aussi une prise
de risque incompressible (4 décès/4 000 procédures dans
une série américaine) avec une espérance de succès pri-
maire supérieure à 90 % et de bon résultat confirmé dans
plus de 75 % des cas, avec 3 ans de recul [9].
Le syndrome du QT long congénital
Le syndrome du QT long congénital (SQTL), clinique-
ment et génétiquement hétérogène, se caractérise par un
allongement de l’intervalle QT, associé à un risque élevé
de survenue de troubles graves du rythme ventriculaire
(torsades de pointe, fibrillation ventriculaire) pouvant
entraîner malaises, syncopes et mort subite. La distinction
historique entre syndrome de Jervell et Lange-Nielsen
(forme récessive avec surdité) et syndrome de Romano-
Ward (forme dominante sans surdité) tend à être aban-
donnée au profit d’une classification s’appuyant sur les
progrès de la génétique moléculaire. Les gènes responsa-
bles des formes LQT1, LQT2, LQT5 et LQT6 codent pour
des canaux potassiques (KCNQ1, HERG, KCNE1, KCNE2
respectivement) et celui responsable de la forme LQT3
code pour le canal sodique cardiaque (SCN5A). La préva-
lence génétique est actuellement estimée à 1/5 000 indi-
vidus [10, 11]. Le phénotype du SQTL est caractérisé par
un allongement de l’intervalle QT (QTc > 440 ms),
mesuré dans les dérivations D2 ou V5 de l’ECG. Ces der-
nières années, le diagnostic génétique est devenu l’une
des méthodes de référence pour le diagnostic de SQTL.
Cependant, la technologie nécessaire n’est pas disponible
partout. L’hétérogénéité génétique et allélique observée
dans le SQTL ainsi que la grande taille de ces gènes ren-
dent difficile son diagnostic génétique. Actuellement
encore, le criblage de ces gènes est effectué par la tech-
nique électrophorétique d’analyse de conformation des
ADN simples brins (SSCP) suivi de l’identification des
Tableau 2. Principaux troubles du rythme pouvant être
responsables de malaises chez le nourrisson
Tachycardie jonctionnelle réciproque paroxystique
Syndrome de Wolff-Parkinson-White
Tachycardie ventriculaire idiopathique
Syndrome de QT long
Bloc auriculo-ventriculaire
Bradycardie vagale paroxystique
A
B
Figure 2. Tachycardie supraventriculaire paroxystique (A) à 280/mm, méconnue depuis 3 jours chez un nourrisson de 6 mois, après un
premier examen médical, évolution défavorable malgré les manœuvres de réanimation. En B) : tracés préagoniques.
mt pédiatrie, vol. 12, n° 2, mars-avril 2009 101
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.

anomalies par séquençage. Par cette technique, le géno-
type n’est établi que dans 60 à 70 % des cas (séries euro-
péennes). L’analyse morphologique de l’onde T sur l’ECG
de sujets génétiquement identifiés montre des aspects
morphologiques assez spécifiques pour les trois princi-
paux gènes connus. Dans la forme LQT3, l’intervalle
QTc est très allongé avec une onde T tardive et de grande
amplitude. Dans la forme LQT2, l’onde T est de faible
amplitude. Dans la forme LQT1 qui est la plus fréquente,
la morphologie de l’onde T est monophasique avec une
base élargie, sensiblement normale, ce qui rend son diag-
nostic plus difficile. L’analyse de l’ensemble des tracés
ECG des membres d’une même famille améliore la sensi-
bilité et la spécificité pour un patient donné. Les anoma-
lies de durée et de morphologie de la repolarisation ven-
triculaire font partie des critères diagnostiques du SQTL.
L’analyse du Holter-ECG est très contributive par rapport
àl’ECG [12], dans la détection d’anomalies de l’onde T
chez des patients suspects de SQTL. Ces anomalies de
l’onde T « en double bosse » présente sur une dérivation
orientent vers une mutation dans HERG, en particulier si
elles sont associées à une inversion de l’onde T dans
l’autre dérivation du Holter. Le mode de déclenchement
des événements rythmiques semble dépendre de la forme
génétique. En effet, le plus souvent le facteur déclenchant
d’un événement cardiaque grave en cas de LQT1 est un
stress, surtout à l’effort. Les patients LQT2 ont eux des
syncopes ou des troubles du rythme survenant plutôt
lors d’une stimulation auditive ou à l’émotion. De plus,
les facteurs de récidives d’évènements cardiaques dépen-
dent du mode de survenue de l’évènement initial.
Les patients LQT1 sont symptomatiques principalement
àl’effort (68 %), les patients LQT2 à l’émotion (51 %) ou
lors du sommeil (34 %) et les patients LQT3 durant le
repos ou le sommeil (53 %), et alors éventuellement res-
ponsable de certains cas de mort subite du nourrisson.
La survenue des symptômes dépend de l’âge et du
sexe. L’âge de survenue du premier événement clinique
est plus précoce chez les garçons que chez les filles.
Cependant, il reste très difficile d’évaluer le pronostic de
cette maladie pour un patient donné. L’incidence des évé-
nements cardiaques est plus importante dans les groupes
LQT1 et LQT2 que dans le groupe LQT3 et augmente
avec la valeur de l’espace QTc, indépendamment du
génotype. La mortalité cardiaque est comparable dans
les 3 groupes mais la létalité des événements est plus éle-
vée dans le groupe LQT3. Ainsi, dans les formes LQTl et
LQT2, les patients sont plus souvent symptomatiques
mais en meurent peu, alors que dans la forme LQT3, les
patients sont moins symptomatiques mais le risque mortel
à chaque syncope est plus important.
Une forme particulièrement grave de QT long avec
bloc est l’apanage du nouveau-né et peut même être res-
ponsable de troubles du rythme anténataux et de mort
subite in utero. Chez ces nouveau-nés au QT parfois
monstrueux, l’existence d’un bloc auriculo-ventriculaire
de degré variable, parfois complet, majore la bradycardie
et les anomalies de la repolarisation (figure 3). La mise en
place d’un stimulateur cardiaque couplé au traitement
bêtabloquant n’est parfois pas suffisant pour éviter l’issue
fatale du fait d’un état de mal rythmique. Ces formes gra-
vissimes se voient surtout chez les sujets porteurs d’un
LQT2 et chez les homozygottes SCN5A. Concernant le
traitement d’urgence, le magnésium (Mg) a fait la preuve
de son efficacité tant expérimentale qu’en clinique. On
utilise 2 grammes (pour 1,73 m
2
), c’est-à-dire 20 mL
d’une solution à 10 % de sulfate de magnésium, en intra-
veineuse en cas de TDP, renouvelable 1 fois 5 à 10 minu-
tes plus tard. Le relais sera pris par une perfusion de Mg de
3 à 10 mg/mn. Avec le magnésium, une supplémentation
potassique doit être donnée pour maintenir un taux
sérique de potassium aux alentours de 4,5 mMoles/L.
Il peut être parfois nécessaire de recourir à la stimulation
cardiaque temporaire. Le traitement de référence dans la
prévention des torrades de pointe est la prescription de
bêtabloquants à tous les patients symptomatiques. En
effet, il y a une vingtaine d’années la mortalité rythmique
spontanée était considérable, de l’ordre de 70 % à 10 ans,
survenant habituellement dans les 2 ou 3 premières
décennies. L’utilisation du traitement bêtabloquant à par-
N
435 440 435 430 425 430 435 440 750 850 850
NNNNNNNN N N N
Figure 3. Nourrisson de 3 mois présentant un syndrome de QT long familial : le rythme cardiaque est tantôt normal à l’auscultation (fré-
quence = 140/mm), tantôt lent par bloc auriculo-ventriculaire intermittent, associé.
mt pédiatrie, vol. 12, n° 2, mars-avril 2009
Origine cardiaque des malaises
102
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 03/06/2017.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%