(PNDS) Délétion 22q11

Centre de Référence Anomalies du Développement
et Syndromes Malformatifs Sud Montpellier
Protocole National de Diagnostic et de Soins (PNDS)
Délétion 22q11
Novembre 2015

2
Sommaire
Liste des abréviations ............................................................................................................... 4
Synthèse à destination du médecin traitant .............................................................................. 5
Texte du PNDS ........................................................................................................................ 8
I/ Introduction ....................................................................................................................... 8
II/ Objectifs du protocole national de diagnostic et de soins ............................................ 8
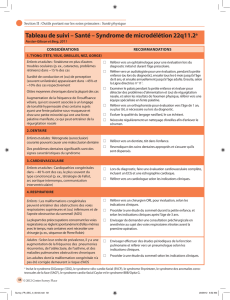
III/ Diagnostic et Évaluation initiale ..................................................................................... 9
Objectifs 9
Professionnels impliqués (et modalités de coordination) 10
Circonstances de découverte / Suspicion du diagnostic 12
Confirmation du diagnostic / diagnostic différentiel 15
Évaluation de la sévérité / extension de la maladie / recherche de comorbidités / évaluation
du pronostic 17
Évaluation de la sévérité / évaluation du pronostic 25
Recherche de contre-indications au traitement 29
Annonce du diagnostic et information du patient 29
Conseil génétique 29
IV/ Prise en charge thérapeutique ....................................................................................... 30
Objectifs 30
Professionnels impliqués (et modalités de coordination) 30
Prise en charge thérapeutique (pharmacologique et autre) 31
Prise en charge par pathologie ........................................................................................ 31
Éducation thérapeutique et modification du mode de vie (au cas par cas) 37
Recours aux associations de patients 37
V/ Suivi .................................................................................................................................. 38
Objectifs 38
Rythme et contenu des consultations 39

3
Annexe 1 .................................................................................................................................. 44
Liste des participants ................................................................................................................ 44
Annexe 2 .................................................................................................................................. 46
Coordonnées des centres de référence, de compétence et des associations de patients ........ 46
Annexe 3 .................................................................................................................................. 48
Arbre décisionnel en vue du diagnostic et de la prise en charge du déficit immunitaire ............ 48
Annexe 4 .................................................................................................................................. 50
Courbes de croissance pour le syndrome de délétion 22q11 ................................................... 50
Annexe 5 .................................................................................................................................. 65
Points forts et faibles dans le développement cognitif .............................................................. 65
Références bibliographiques .................................................................................................... 656
Participants .............................................................................................................................. 106

4
Liste des abréviations
ACPA Analyse Chromosomique sur Puces à ADN
ALD Affection de Longue Durée
AMM Autorisation de Mise sur le Marché
ATP Atrésie Pulmonaire
CAo Coarctation de l'aorte
CIA Communication InterAuriculaire
CIV Communication InterVentriculaire
CPDPN Centre Pluridisciplinaire de Diagnostic PréNatal
DPI Diagnostic Pré-Implantatoire
DPN Diagnostic PréNatal
FéCLAD Fédération des Centres Labellisés Anomalies du Développement et
Syndromes Malformatifs
FISH Fluorescent In Situ Hybridization (Hybridation In Situ Fluorescente)
HAS Haute Autorité de santé
IAA Interruption de l’Arc Aortique
IVP Insuffisance Vélo-Palatine
Mb Mégabase
MCC Malformation Cardiaque Conotroncale
MLPA Multiplex Ligation-dependent Probe Amplification
PCR Polymerase Chain Reaction (Réaction de polymérisation en chaîne)
PNDS Protocole National de Diagnostic et de Soins
QI Quotient Intellectuel
SDG Syndrome de Di George
SVCF Syndrome Vélo-Cardio-Facial
TAC Tronc Artériel Commun
TDAH Troubles Déficitaires de l’Attention avec Hyperactivité
TF Tétralogie de Fallot

5
Synthèse à destination du médecin traitant
La délétion 22q11 (ou microdélétion 22q11.2) est une anomalie du développement rare qui
associe :
Des cardiopathies congénitales, plus particulièrement conotrocales (CIV, Tétralogie
de Fallot, atrésie pulmonaire, tronc artériel commun, interruption de l’arc aortique,…),
Des anomalies du palais (insuffisance vélo-pharyngée, fente palatine ou labio-
palatine),
Des hypocalcémies, surtout en période néonatale,
Un déficit immunitaire,
Des difficultés d’apprentissage et particulièrement des troubles du langage,
Un aspect particulier du visage,
Et moins fréquemment:
Des troubles digestifs avec dysphagie et reflux gastro-œsophagien,
Des anomalies génito-urinaires,
Des anomalies squelettiques,
Un déficit en hormone de croissance,
Des maladies auto immunes,
Un déficit auditif,
Des troubles ophtalmologiques,
Des anomalies dentaires,
Un profil cognitif particulier caractérisé par des difficultés de la mémoire visuelle
immédiate et des fonctions exécutives,
Des troubles psychologiques pendant l’enfance (hyperactivité, troubles de l’attention,
…),
Une évolution à partir de l’adolescence et à l’âge adulte vers des pathologies
psychiatriques (schizophrénie en particulier).
Plus de 180 anomalies ont été répertoriées chez les personnes porteuses d’une
délétion 22q11.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
1
/
108
100%