DocCours Chimie Organique : réactions

DocCours Chimie Organique : réactions orgaII - 1/12
Figure 1 : Quelques facteurs contrôlant une réaction
Contrôle de charge
L’espèce A est pauvre en e- (électrophile), B est riche en e- (nucléophile). A et B s’attirent et vont réagir pour
former un nouveau composé : A+ + B- A-B
Contrôle orbitalaire (2ème année)
Conditions pour que la réaction se fasse :
les orbitales moléculaires des composés A et B doivent être :
a) en phase, b) proches en énergie, et c) de symétries "compatibles"
Contrôle stérique
Le réactif A aura plus de mal à rejoindre
le site + encombré (à gauche) que le site + de droite.
On parle d’encombrement stérique.
En première année, les réactions seront guidées par le contrôle de charge, et dans une moindre mesure, par
l’encombrement stérique.
On prendra donc grand soin d’étudier la répartition des charges des composés avant d’écrire les réactions.
BCA réaction possible
entre A et C,
pas entre A et B
OA "s" OA "pz"OA "pz"
+
-
+
-
+
gêne ou encombrement
stérique
Me
Me
Me
+A
-
approche
difficile
H
HH
+
approche
facile
réaction possible
entre A et C
Figure 2 : Classification des réactifs : Acides et Bases -Nucléophiles et Electrophiles
Acide (Définitions au sens de Bronsted)
Toute molécule pouvant libérer un H+ : AH → A- + H+ (A- est la base conjuguée)
Exemples :
Base (Définitions au sens de Bronsted)
Toute molécule pouvant capturer un H+ : B + H+→ BH+ (BH+ est l’acide conjugué)
Exemples :
Nucléophile
Toute espèce qui a une affinité pour les "noyaux", c'est-à-dire les centres chargés positivement.
Concrètement : toute molécule possédant au moins un doublet riche en électron (DNL ou nuage ).
Un nucléophile n’est pas forcément chargé < 0.
Exemples :
Electrophile
Toute espèce ayant une affinité pour les "électrons" c'est-à-dire les centres chargés négativement.
Concrètement : toute molécule ayant une charge > 0, une lacune électronique, ou un atome déficitaire en
électrons. Un électrophile n’est pas forcément chargé > 0.
Exemples :
R O
O
H
ROH
S
H H
HH
O
H H NH
H
H
H
H2OH2S ROH RCOOH NH4+
RO
S
H
O
HR O
O
NH
H
NH
H
H
HO-HS-RO-RCOO- NH3NH2-
H2OROH
ROH
O
H H
HS-RCOO-
S
HR O
ONH
H
H
NH3
HO-
O
H
H2S
S
H H carbanion
RO
RO-
CH
H
H
alcène
X = halogène
HCH
H
H
proton
CH
H
H
carboradical
X
carbocation

DocCours Chimie Organique : réactions orgaII - 2/12
Figure 3 : Différence entre basicité et nucléophilie
Le concept nucléophile/électrophile est un concept de cinétique, et est donc lié aux vitesses de réaction.
Le concept acide/base un concept de thermodynamique, et est donc lié aux équilibres de réactions.
Exemple : OH- et SH- ont une structure similaire, et sont tous deux basiques et nucléophiles.
Concept acide/base (aspect thermodyamique)
pKa (H2O/OH-) = 14 ; pKa (H2S/SH-) = 7
Donc l’acide H2S est plus fort que l’acide H2O
et la base SH- est plus faible que la base OH-
OH- pourra plus facilement que SH-, arracher un H+ à un même site acide.
Concept nucléophile/électrophile (aspect cinétique)
Pourtant, le doublet non liant sur S est plus gros que celui de O,
car les électrons de ce doublet ont un nombre quantique n = 3
(couche de valence), contre n = 2 pour O.
Ils sont donc plus éloignés du noyau et plus disponible pour établir
une liaison avec un autre atome.
Par conséquent SH- est meilleur nucléophile que OH-
SH- réagira plus vite que OH- sur un même site électrophile.
HO-
O
HS
H
HS-
Figure 4 : Conventions d’écriture des mouvements électroniques
Rappel : Les mouvements électroniques sont décrits par des flèches précises :
flèche à deux pointes (ou dents) flèche à une « pointe »
= mouvement de deux électrons = mouvement d’un électron
Ces flèches représentent le mouvement des électrons :
elles partent donc des zones riches en électrons (doublet non liant, doublet p d’une double liaison, …)
et vont vers des zones plus pauvres en électrons (atome portant une charge +, radical libre, …)
Exemples :
cassure d'une liaison
OH
H+
-La flèche part du milieu de la liaison qui se casse
et va vers l'atome O.
Un nouveau doublet non liant est créé
OH +H
OH +HOH
H
La flèche part du doublet non liant qui va réagir,
(et non de la charge formelle - sur O),
et va vers l'atome H (et non sa charge formelle +)
Une nouvelle liaisonest créée.
formation d'une liaison à partir d'un doublet non liant
formation d'une liaison à partir d'un doublet d'une double liaison
La flèche part du milieu du doublet qui va réagir,
et va vers l'atome H (et non sa charge formelle +)
Une nouvelle liaisonest créée, invisible en
notation topologique, entre un des carbones de C=C
et H.
Cela donne deux carbocations possibles, selon que
H se fixe sur le C de gauche (cas B) ou de droite (cas A).
Bien noter que la charge formelle + est sur le C qui n'a
pas reçu le H.
+H
mélange non équimolaire
AB
Figure 5 : Les deux modes de rupture d’une liaison
Cassure hétérolytique :
Cassure homolytique :
+
A
cassure hétérolytique
(apparition de charge) ions
A B B
CH
H
HCl CH
H
H
carbocation
+ Cl CH
H
HLi + Li
CH
H
H
carbanion ion lithium
+
cassure homolytique
(pas d'apparition de charge) A B
radicaux libres
A B
CH
H
HCl
carboradical
+ Cl
CH
H
H
atome Cl

DocCours Chimie Organique : réactions orgaII - 3/12
Figure 6 : Classification des réactions (de 1ère année)
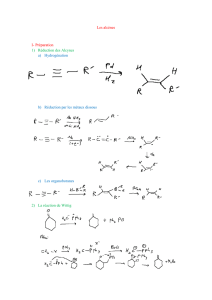
Substitution :
Symbole de la réaction : il dépend du réactif A approchant le substrat BC
SN : substitution nucléophile si A est un nucléophile
SE : substitution électrophile si A est un électrophile
Addition :
Symbole de la réaction
AN:addition nucléophile ; si le 1er atome s’approchant de C=D est un nucléophile (il faut voir le mécanisme
pour le savoir)
AE : addition électrophile ; si le 1er atome s’approchant de C=D est un électrophile
A + B-C A-B + C
substitution de C par A
sur l'atome B
A-B + C=D C-D
addition
de AB sur C=D
A B
Figure 7 : Sélectivité d’une réaction
Régiosélectivité
Une réaction est dite régiosélective si elle privilégie
la formation d’un isomère de constitution B par
rapport à un autre isomère C.
Exemple :
Stéréosélectivité
Une réaction est dite stéréosélective si elle privilégie la
formation d’un stéréoisomère B par rapport à un autre B’
(voir exemple plus bas)
Stéréospécificité
Une réaction est dite stéréospécifique si
elle est stéréosélective
et qu’un stéréoisomère A’ de A donne B et B’
dans les proportions inverses.
Si B et B’ sont énantiomères, la réaction est
dite énantiospécifique.
Si B et B’ sont diastéréoisomères, la réaction est
dite diastéréospécifique.
Exemple :
B et C sont des
isomères
de constitution
B et B’ sont des
stéréoisomères
A et A’ sont des
stéréoisomères
B et B’ sont des
stéréoisomères
Cl + Br-solvant
peu polaire
Br Br
++ Cl-
composé S
minoritaire : 10 %
composé S composé R
majoritaire: 90 %
Cl + Br-Br Br
++ Cl-
composé S
majoritaire 90 %
composé R composé R
minoritaire: 10 %
réaction stéréosélective
réaction
stéréospécifique
ici énantiospécifique
car les deux composés
obtenus (R et S) sont énantiomères
solvant
peu polaire
+ HCl
Cl Cl
+
minoritaire : 18%
solvant polaire
majoritaire: 82%
+ HCl
Cl Cl
+
solvant apolaire
majoritaire : 76% minoritaire: 24%

DocCours Chimie Organique : réactions orgaII - 4/12
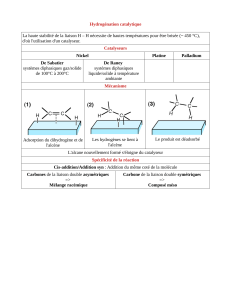
Figure 8 : Réactivité des alcènes
Structure électronique de la liaison C=C
(cas de l’éthène)
Les deux carbones sont AX3E0
La géométrie est donc triangulaire plane
avec des angles de base 120°.
La double liaison occupe une place plus importante, donc l’angle HCH se resserre.
Il y a 5 liaisons à symétrie axiale notées
et une liaison à symétrie plane notée ,
perpendiculaire au plan de la molécule,
et obtenue par interaction des deux OA
2pz des carbones (les signes ne sont pas
des charges, mais ceux des OA)
C’est cette liaison qui empêche la libre rotation
de C=C, d’où l’isomérie Z/E.
Stabilité de la liaison
On note que E(C=C) > E(C-C)
Mais : E(C=C) < 2 E(C-C)
La liaison double C=C, constituée d’une liaison et
d’une liaison , n’est pas aussi forte que 2 liaisons simples (liaison ).
L'approximation E(C=C) = E() + E()E() + E(C-C) < 2 E(C-C) conduit à : E() < E()
La liaison est plus fragile que la liaison
La grande réactivité des alcènes est donc principalement contenue dans leur liaison , car :
elle est fragile
elle s’étale loin dans l’espace
elle est très polarisable (déformable)
elle est riche en électrons disponibles (comme un dnl)
Réactivité des alcènes
(réactions de 1ère année)
les OA 2pz
interagissent
et forment la
liaison
C C
H
H
H
H
C
H
H
CH
H
117° 121,5°
+
+
-
-
+
-
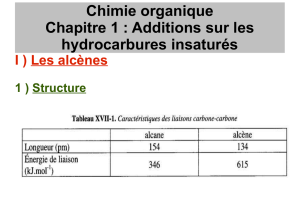
C-H
C-C
C=C
Longueur de liaison (pm)
110
154
134
Energie de liaison (kJ mol-1)
414
348
615
les alcènes sont très réactifs (par rapport aux
alcanes) et se comportent comme des nucléophiles
les alcènes subissent des additions (ouverture de C=C par cassure de la liaison )
Cassure des liaisons ET sous
l’action d’un oxydant comme l’ozone O3
Réaction
d’ozonolyse
C
H
H
CH
H
E
(électrophile = H+, X∙, …)
Cassure de la liaison
(fragile et nucléophile)
à l’approche d’un électrophile E
Réactions d’addition
électrophile AE
(partie la plus importante)
Figure 9 : Stabilité des alcènes
Les alcènes n'ont pas tous la même stabilité ni la même
réactivité. Une méthode pour étudier la stabilité des alcènes
est de mesurer leur énergie d’hydrogénation H° (énergie
libérée au cours de l’addition d’une mole de H2).
Les trois alcènes du graphique conduisent tous au même
alcane (le butane), niveau de référence dans le graphe.
Plus H° est grande, moins l’alcène de départ est stable.
On retiendra que :
la stabilité d’un alcène augmente avec le nombre de groupes portés par la double liaison
(les groupes apportent des électrons par effet +I à la liaison C=C insaturée)
les alcènes sont également stabilisés par les effets +M (doubles liaisons conjuguées)
un alcène cis est moins stable qu’un alcène trans (encombrement stérique)
1 cal = 4,18 J
Ep
A : doubles liaisons
conjuguées
B moins stable
que A

DocCours Chimie Organique : réactions orgaII - 5/12
Figure 10 : Additions électrophiles (AE) sur la double liaison C=C - Généralités
Réaction générale :
Problème de régiosélectivité ?
Si l’alcène de départ et le composé AB sont tous les deux dissymétriques, on peut obtenir un mélange de deux
produits, isomères l’un de l’autre, selon que A se fixe sur le carbone de droite ou de gauche.
Cette réaction peut donc être ou non régiosélective.
C’est l’expérience et l’étude du mécanisme qui dira si elle l’est vraiment.
Problème de stéréosélectivité ?
S’il y a apparition d’un carbone asymétrique, la réaction peut produire plusieurs stéréoisomères.
Elle peut donc être ou non stéréosélective (ou stéréospécifique)
Là encore c’est l’expérience et l’étude du mécanisme qui dira si elle l’est vraiment.
C C + A B
+ C C
AB
C C
BA
+
mélange
r
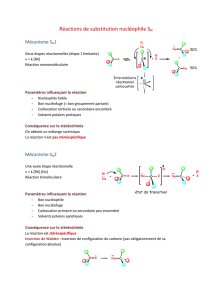
Figure 11 : Additions électrophiles ioniques –Hydrohalogénation - Bilan
Il s’agit de l’addition ionique de HX sur C=C (X=halogène : F, Cl, Br ,I).
On obtient un mélange de dérivés halogénés.
Bilan :
Mode opératoire : la réaction se fait
à température élevée (170 °C) en phase gazeuse pour les premiers alcènes (peu réactifs)
et à froid pour les alcènes plus lourds, HX gazeux barbotant dans l'alcène pur et
liquide, ou bien l'alcène et HX étant dissouts dans un solvant polaire.
Il y a dégagement de chaleur (réaction exothermique).
Régiosélectivité expérimentale : Règle de Markovnikov (1869)
Lors de l’addition électrophile ionique d’un composé H+─A- sur un alcène dissymétrique, A se fixe sur le
carbone le plus substitué.
Stéréosélectivité expérimentale : aucune
C C
R+H X
+ C C
XH
R
C C
HX
R
+
mélange
Figure 12 : Additions électrophiles ioniques –Hydrohalogénation - Mécanisme
Mécanisme en solvant polaire
Mécanisme par stades en deux étapes.
1ère étape : étape lente (la plus difficile)
La liaison (nucléophile), capture le proton H+
(électrophile) de l’acide H-X. On obtient deux carbocations.
2ème étape :
Les 2 carbocations sont attaqués par le nucléophile X⊖
On obtient deux composés A et B régioisomères
(voir la suite pour la détermination du composé majoritaire)
C C
H
R
+ X C C
H
R
X
composé A
+ X C C
X
R
H
C C
H
R
composé B
+ C C
H
R
C C
H
R
+
H X
+C C
R
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%