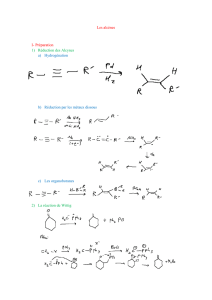
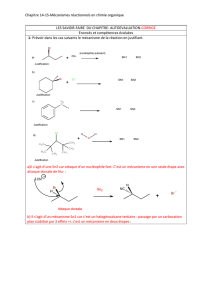
ORGAII - Le site de la Sup 1

page 1
Chimie Organique
Partie II
Réactions

page 2
Généralités
Contrôle d’une réaction
Classification des réactifs
Conventions d’écriture
des mouvements électroniques
Mode de coupure d’une liaison
Classification des réactions
Sélectivité d’une réaction

page 3
Contrôle de charge
L’espèce A est pauvre en e-, B est riche en e- (présence d’un dnl par exemple). A et B
s’attirent et vont réagir pour former un nouveau composé : A++ B-A-B
Contrôle orbitalaire (2ème année)
Conditions pour que la réaction se fasse :
les orbitales doivent être en phase, proches
en énergie, et de symétries « compatibles ».
Contrôle stérique
Le réactif A aura plus de mal à rejoindre
le site +encombré (à gauche)
que le site +de droite.
On parle d’encombrement stérique.
Principaux facteurs contrôlant une réaction
En première année, les réactions seront guidées par le contrôle de charge, et dans une
moindre mesure, par l’encombrement stérique.
On prendra donc grand soin d’étudier la répartition des charges des composés avant
d’écrire les réactions.
gêne ou encombrement
stérique
Me
Me
Me
+A
-
approche
difficile
H
HH
+
approche
facile
réaction possible
entre A et C
BCA réaction possible
entre A et C,
pas entre A et B
OA "s" OA "pz"OA "pz"
+
-
+
-
+

page 4
Définitions au sens de Bronsted
Acide
Toute molécule pouvant libérer un H+: AH →A-+ H+ (A-est la base conjuguée)
Quelques exemples (mettre en couleur le H acide)
Base
Toute molécule pouvant capturer un H+: B + H+→ BH+(BH+est l’acide conjugué)
Quelques exemples (mettre en couleur le site basique)
Classification des réactifs
Acides et bases
RO
S
H
O
HR O
O
NH
H
NH
H
H
HO-HS-RO-RCOO-NH3NH2-
R O
O
H
ROH
S
H H
HH
O
H H NH
H
H
H
H2OH2S ROH RCOOH NH4+

page 5
Nucléophile
(traduction : qui a une affinité pour les « noyaux » = centres chargés positivement)
Concrètement : toute molécule possédant au moins un doublet non liant (riche en électron).
Un nucléophile n’est pas forcément chargé <0.
Quelques exemples (mettre en couleur le site nucléophile)
Electrophile
(traduction : qui a une affinité pour les « électrons » = centres chargés négativement)
Concrètement : toute molécule ayant une charge >0, une lacune électronique, un atome
déficitaire en électrons. Un électrophile n’est pas forcément chargé >0.
Quelques exemples (mettre en couleur le site électrophile)
Classification des réactifs
Nucléophiles et électrophiles
X = halogène
HCH
H
H
proton
CH
H
H
carboradical
X
carbocation
H2OROH
ROH
O
H H
HS-RCOO-
S
HR O
ONH
H
H
NH3
HO-
O
H
H2S
S
H H carbanion
RO
RO-
CH
H
H
alcène
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
1
/
47
100%