ABREVIATIONS

ABREVIATIONS
ACFA : Arythmie cardiaque par fibrillation auriculaire
Ao : Aorte
ASCD : Artère sous-clavière droite
ASCG : Artère sous-clavière gauche
BBD : Bloc de branche droit
Bpm : Battements par minute
CA : Canal artériel
CAV : Canal atrio-ventriculaire
CIA : Communication inter-auriculaire
CIV : Communication inter-ventriculaire
CMG : Cardiomégalie
CoA : Coarctation de l’aorte
Cpm : Cycles par minute
ECG : Electrocardiogramme
FC : Fréquence cardiaque
FR : Fréquence respiratoire
HAG : Hypertrophie auriculaire gauche
HMG : Hépatomégalie
HTA : Hypertension artérielle
HTAP : Hypertension artérielle pulmonaire
HVD : Hypertrophie ventriculaire droite
HVG : Hypertrophie ventriculaire gauche
IC : Insuffisance cardiaque

ICT : Index cardiothoracique
KT : Cathétérisme cardiaque
PGE1 : Prostaglandine E1
RM : Rétrécissement mitral
TA : Tension artérielle
TABC : Tronc artériel brachio-céphalique
TGV : Transposition des gros vaisseaux
VCI : Veine cave inférieure
VD : Ventricule droit
VG : Ventricule gauche

PLAN
Première partie :
Introduction ………………………………………………………………………………. 3
Historique …………………………………………………………………………………. 5
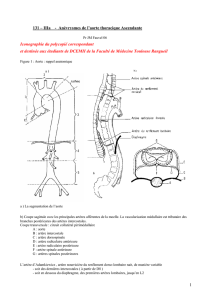
Anatomie de l’aorte ……………………………………………………………………… 7
A- Aorte thoracique…………………………………………………………………. 7
B- Aorte abdominale………………………………………………………………… 9
Epidémiologie ……………………………………………………………………………… 10
Physiopathologie …………………………………………………………………………. 11
Formes du nouveau-né…………………………………………………………….. 12
Formes de l’adulte…………………………………………………………………… 13
Formes isolées……………………………………………………………………….. 13
Formes complexes…………………………………………………………………… 14
Physiopathologie de l’HTA………………………………………………………… 14
Conséquences de la coarctation…………………………………………………. 15
Anatomopathologie ……………………………………………………………………… 17
A- La coarctation aortique………………………………………………………… 17
B- Formes selon le siège de la sténose………………………………………… 18
1- Localisations isthmiques………………………………………………… 18
2- Localisations atypiques………………………………………………….. 18
C- Lésions cardiovasculaires associées………………………………………… 19
Embryologie ……………………………………………………………………………….. 20
Rappel sur la circulation fœtale…………………………………………………… 20
Modifications circulatoires postnatales…………………………………………. 22

Développement des vaisseaux……………………………………………………. 23
Etiopathogénie ……………………………………………………………………………. 24
A- Théorie hémodynamique………………………………………………………. 24
B- Théorie mécanique……………………………………………………………….. 24
C- Autres hypothèses……………………………………………………………….. 25
Diagnostic anténatal ……………………………………………………………………… 26
Etude clinique ……………………………………………………………………………… 29
A- Les formes des nouveaux-nés………………………………………………… 29
1- Circonstances de découverte……………………………………………. 29
2- Examen physique…………………………………………………………… 30
B- La coarctation de l’aorte du grand enfant…………………………………… 33
1- Circonstances de découverte……………………………………………. 33
2- Examen physique…………………………………………………………… 33
C- Formes anatomiques……………………………………………………………. 35
1- Forme peu serrée…………………………………………………………… 35
2- Forme étendue……………………………………………………………… 35
3- La coarctation abdominale…………………………..…………………... 35
D- Formes associées à d’autres anomalies……………………..……………… 36
1- Anomalie de l’orifice aortique…………………………………………… 36
2- Les shunts associés………………………………………………………… 36
3- Anomalies de naissance des artères sous-clavières……………….. 36
E- CoA et grossesse………………………………………………………………… 36
Paraclinique ……………………………………………………………………………….. 37
A- ECG…………………………………………………………………………………. 37
1- Chez le nouveau-né………………………………………………………. 37
2- Chez le grand enfant……………………………………………………… 37
B- Radiographie thoracique………………………………………………………… 38

1- Chez le nouveau-né……………………………………………………….. 38
2- Chez le grand enfant………………………………………………………. 38
C- Echographie transthoracique…………………………………………………. 43
1- Echographie en mode TM………………………………………………… 43
2- Echographie bi-dimentionnelle…………………………………………. 44
3- Echographie doppler………………………………………………………. 44
D- IRM………………………………………………………………………………….. 48
E- Cathétérisme et angiographie………………………………………………… 52
Evolution et pronostic …………………………………………………………………… 54
A- Evolution selon l’âge de découverte………………………………………… 54
B- Evolution selon les formes anatomocliniques……………………………… 56
Traitement …………………………………………………………………………………. 57
A- Moyens……………………………………………………………………………… 57
1- Traitement médical………………………………………………………… 57
1.1- Traitement de la défaillance…………………………………… 58
1.2- Prostaglandine……………………………………………………. 71
2- Traitement chirurgical……………………………………………………… 72
Généralités……………………………………………………………….. 72
Abord de la coarctation……………………………………………….. 73
Technique de Crafoord : résection anastomose…………………. 75
Technique de Waldhausen : plastie anastomose………………… 81
Résection étendue…………………………………………………….. 83
Résection et tube………………………………………………………. 83
Plastie d’élargissement prothétique……………………………….. 84
Tube aortique latéral………………………………………………….. 84
Résection suture par sternotomie médiane………………………. 84
3- Angioplastie percutanée……….……………………………………….… 85
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
1
/
196
100%