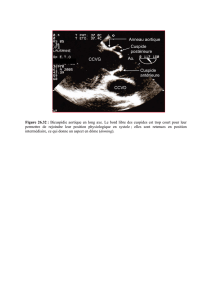
Nous rapportons 2 observations d`hypoplasie du cœur gauche dont

L’hypoplasie du coeur gauche est une cardiopathie congénitale qui, bien que sporadique, est extrêmement sérieuse.
Ce
terme
sert
à
désigner
des
insuffisances
de
développement
du
ventricule
gauche,
de
ses
valves
d'admission
et
de
H.Mahfoudhi, H.Ben Salem*, I.Hamdi, Gh.Chniti, S.Blibech*, Th.Filali, I.Saaïdi, B.Jdaïda, A.Haggui, R.Dahmen, N.Hajlaoui,
D.Lahidheb, W.Fehri, A.Douagi H.Haouala
Service de cardiologie-Hôpital Militaire- Tunis
*Service de pediatrie- Hôpital Militaire- Tunis
Ce
terme
sert
à
désigner
des
insuffisances
de
développement
du
ventricule
gauche,
de
ses
valves
d'admission
et
de
sortie, ainsi que de l'aorte ascendante. Le degré et le groupement des lésions varient notablement d'un cas à l'autre
si bien que la malformation porte souvent le nom de sa lésion prédominante. L'espérance de vie est ainsi
intimement liée à la durée de perméabilité du canal artériel.
Nous rapportons 2 observations d’hypoplasie du cœur gauche dont le diagnostic a été fait en période
néonatale grâce a l’échocardiographie faite devant un allongement du temps de recoloration avec tachypnée
dans le premier cas et devant l’apparition et l'accentuation rapide d'une cyanose dans le 2ème cas.
L’évolution était rapidement fatale dans les deux cas dans un tableau de défaillance cardiaque sévère juste
après
la
fermeture
du
canal
artériel
.
après
la
fermeture
du
canal
artériel
.

• Le syndrome d'hypoplasie du coeur gauche (SHCG) désigne une anomalie de
développement des structures gauches du coeur, obstruant la circulation sanguine
dans les vaisseaux issus du ventricule gauche.
• Ce syndrome est également caractérisé par un développement insuffisant du ventricule
gauche, de l'aorte et de l'arc aortique, ainsi que par une atrésie ou une sténose mitrale.
• Le SHCG concerne environ 1 à 2 naissances d'enfants vivants sur 6250.
• Les nouveau-nés atteints naissent généralement à terme et en bonne santé. Lorsque le
canal artériel se ferme, la perfusion systémique diminue, entraînant une hypoxie, une
acidose et un choc cardiogénique. Généralement, on détecte un souffle cardiaque non
spécifique
ou
l'absence
de
souffle
.
Le
deuxième
bruit
cardiaque
est
fort
et
unique
en
spécifique
ou
l'absence
de
souffle
.
Le
deuxième
bruit
cardiaque
est
fort
et
unique
en
raison de l'atrésie aortique. Le volume du foie est souvent augmenté, suite à
l'insuffisance cardiaque congestive.
• Comme pour la plupart des cardiopathies congénitales, la cause embryologique de ce
syndrome n'est pas entièrement connue.
• L'échocardiogramme est la méthode diagnostique de choix. Le syndrome peut être
diagnostiqué par une échocardiographie foetale entre 18 et 22 semaines de grossesse.
Le diagnostic différentiel inclut les autres lésions obstructives gauches pour lesquelles
la circulation systémique dépend de la perméabilité du canal artériel (sténose aortique
critique, coarctation de l'aorte, interruption de l'arc aortique, voir ces termes).

• L'échocardiogramme est la méthode diagnostique de choix. Le syndrome peut être
diagnostiqué par une échocardiographie foetale entre 18 et 22 semaines de grossesse. Le
diagnostic différentiel inclut les autres lésions obstructives gauches pour lesquelles la
circulation systémique dépend de la perméabilité du canal artériel (sténose aortique
critique, coarctation de l'aorte, interruption de l'arc aortique, voir ces termes).
• Les nouveau-nés atteints de ce syndrome doivent bénéficier d'une chirurgie néonatale,
puisque leur circulation systémique est canal-dépendante. Actuellement, il y a deux options
thérapeutiques principales : la transplantation cardiaque primaire ou une série
thérapeutiques principales : la transplantation cardiaque primaire ou une série
d'interventions palliatives permettant le fonctionnement cardiaque avec un ventricule
unique. Le choix du traitement dépend des préférences de l'établissement et de
l'expérience du chirurgien. Bien que le taux de survie suivant la première intervention
chirurgicale ait considérablement augmenté au cours des 20 dernières années, une
mortalité et une morbidité importantes restent associées aux deux options thérapeutiques.
Ainsi, les pédiatres cardiologues restent confrontés à des discussions délicates avec les
familles concernant le choix du traitement et le pronostic à long terme, d'autant que
l'information sur la survie à long terme et la qualité de vie des enfants atteints de ce
syndrome est limitée.
1
/
3
100%