LES INDICATIONS DU DOSAGE DES HORMONES STEROIDES

Les indications du dosage des hormones stéroïdes
225
Revue de l'ACOMEN, 1998, vol.4, n°3
Durant les vingt dernières années, les progrès de
limmunoanalyse ont permis dobtenir des anticorps spé-
cifiques dirigés contre de nombreux haptènes et en parti-
culier contre les hormones stéroïdes et leurs précurseurs.
La mise au point de dosages performants a largement con-
tribué à la connaissance de la pathologie surrénalienne et
gonadique. Ces dosages ont, maintenant, complètement
supplanté la mesure très approximative des métabolites
urinaires des hormones sexuelles ou surrénaliennes.
Après un bref rappel sur la biosynthèse des hormones
stéroïdes, nous nous proposons de passer en revue dans
leurs grandes lignes les principales circonstances dans
lesquelles le dosage des hormones stéroïdes et de cer-
tains de leurs précurseurs peut apporter une aide au dia-
gnostic des dysfonctionnements ovariens, testiculaires ou
cortico-surrénaliens.
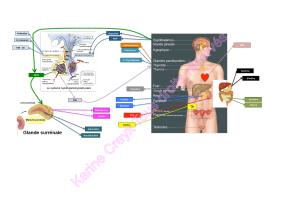
1. La biosynthèse des hormones
stéroïdes
Les hormones stéroïdes dérivent toutes du cholestérol. A
partir de la prégnènolone, précurseur initial, les voies de
synthèse, prioritaires ou exclusives, dépendent de léqui-
pement enzymatique des cellules.
Les principales voies de synthèse des hormones cortico-
surrénaliennes et gonadiques sont schématisées dans la
Figure 1.
Les deux voies horizontales supérieures sont spécifiques
de la cortico-surrénale. Laldostérone est physiologique-
ment produite, exclusivement, par la zone glomérulée et le
cortisol par la zone fasciculée.
La synthèse des androgènes passe préférentiellement par
la voie ∆5 dans la zone réticulée de la cortico-surrénale,
pour aboutir à la production dandrogènes faibles, la
déhydroépiandrostérone (DHEA) et surtout son sulfate
(S-DHEA) environ mille fois plus abondant.
Dans les gonades au contraire, cest la voie D4 qui est
prédominante et qui conduit à la testostérone. Au niveau
de lovaire, lintervention dune aromatase permet la trans-
formation de la testostérone en estradiol.
Il faut remarquer que certaines enzymes sont communes à
différentes voies, ce qui permet de comprendre la traduc-
tion clinique des déficits enzymatiques.
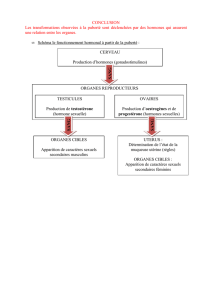
2. Exploration de la fonction ovarienne
De lenfance à la ménopause, le dosage des hormones
sexuelles trouve sa place à tous les âges dans lexplora-
tion des dysfonctions de laxe hypophyso-ovarien.
2.1. Rappel physiologique
La Figure 2, empruntée à Emperaire et Ruffié (1), résume
les différentes étapes de lévolution hormonale au cours
du cycle menstruel : lorsque, sous laction de la FSH, le
follicule sélectionné a atteint un volume et une capacité
sécrétoire suffisants, la brusque augmentation de la pro-
duction destradiol entraîne une stimulation de la sécré-
tion de LH, elle-même responsable de lovulation.
LES INDICATIONS DU DOSAGE
DES HORMONES STEROIDES
B. ARGEMI
Immunotech
- Marseille -
-
- FIGURE 2 -
Evolution hormonale au cours du cycle (d'après Emperaire J.C.
et Ruffié A. (1) avec l'autorisation de l'éditeur

B. ARGEMI
226 Revue de l'ACOMEN, 1998, vol.4, n°3
- FIGURE 1 -
Schéma général de la biosynthèse des hormones stéroïdes chez lhomme.

Les indications du dosage des hormones stéroïdes
227
Revue de l'ACOMEN, 1998, vol.4, n°3
Au cours de la phase lutéale, le corps jaune, principale-
ment stimulé par la LH, produit de la progestérone et de
lestradiol.
Lensemble de ces phénomènes est sous la dépendance
de la sécrétion pulsatile de la "Gonadotrophin Releasing
Hormone" (GNRH) dorigine hypothalamique, soumise au
rétro-contrôle négatif de lestradiol et de la progestérone.
2.2. Les troubles de la puberté
2.2.1. La puberté précoce
Lapparition dune pilosité pubienne et dun début de dé-
veloppement mammaire chez une enfant âgée de moins de
7 ans doit faire évoquer une puberté précoce. Celle-ci est
confirmée par le dosage de FSH. Le degré de maturation
ovarienne est évalué par la mesure de lestradiol sérique et
léchographie. Lensemble du contexte clinique et
paraclinique permet de préciser létiologie (tumeur hypo-
thalamique, syndrome de Mac Cune-Albright, puberté pré-
coce idiopathique) et de mettre en route le traitement ap-
proprié (2).
2.2.2. Les retards pubertaires
En labsence de signes cliniques de développement pu-
bertaire à lâge de 12 ans, et de règles à 15 ans, les dosages
des stéroïdes sériques sintègrent dans lensemble des
investigations qui vont explorer en premier lieu
ladrenarche dont le premier stigmate biologique est lélé-
vation du S-DHEA ou de la DHEA. Le dosage de lestradiol,
comme précédemment associé à léchographie, apprécie le
degré de maturation ovarienne. Lorientation étiologique
est donnée par le dosage de FSH (plus accessoirement de
LH) dont le taux élevé oriente vers une dysgénésie ova-
rienne ; inversement, une valeur basse évoque une origine
haute, et la réponse à la stimulation par GNRH permet daf-
firmer la simple puberté différée.
2.3. Chez la femme en période dactivité
génitale
2.3.1. Exploration des perturbations du cycle menstruel
Le dosage de lestradiol aux différentes phases du cycle et
celui de la progestérone dans la deuxième période du cycle
sont très utiles, associés à létude de la courbe thermique,
pour apprécier limportance dune dysovulation.
Dans le cadre de lexploration dune aménorrhée secon-
daire, le dosage de lestradiol sérique permet dévaluer le
degré de carence estrogénique.
2.3.2. Procréation médicalement assistée (P.M.A)
Le dosage régulier de lestradiol sérique et léchographie
sont les deux éléments de surveillance de la stimulation
ovarienne : lovulation est déclenchée par injection de
gonadotrophine chorionique, ou la ponction ovarienne
réalisée lorsquun ou plusieurs follicules ont atteint un
diamètre de 20 à 22 mm et que lestradiol sérique se situe
entre 500 et 1000 pg/ml (3).
Le dosage de la progestérone en fin de stimulation aurait
une certaine valeur pronostique : son élévation pré-
ovulatoire est corrélée avec léchec de limplantation des
embryons (4).
Dans les syndromes de basse réserve ovarienne, respon-
sables dinfertilité et déchec de la fécondation in vitro, un
taux destradiol supérieur à 80 pg/ml au 3ème jour du cycle,
associé à la classique augmentation du taux de FSH, repré-
sente un élément péjoratif de poids (5).
2.3.3. Au cours de la grossesse
Le dosage de lestriol en fin de grossesse est de moins en
moins utilisé pour rechercher la souffrance ftale, car les
progrès du monitorage ont permis un diagnostic beau-
coup plus précoce de celle-ci.
Par contre, le dosage de lestriol libre est utilisé par cer-
tains laboratoires en association avec celui de lHCG et
éventuellement de lalphafoetoprotéine (AFP) pour éva-
luer, entre la 15e et la 17e semaine daménorrhée, le risque
de trisomie 21 (6).
2.4. La ménopause
En dehors du dosage de la FSH qui permet de confirmer
linstallation de la ménopause, les dosages hormonaux ne
sont utiles ni pour poser lindication dun traitement hor-
monal substitutif (THS), ni pour adapter la dose
destrogènes.
Le dosage de lestradiol et/ou de lestrone sérique nest
utile que chez les femmes présentant une contre-indica-
tion relative au THS et chez lesquelles la persistance dune
production résiduelle destradiol, ou destrone (à partir de
la DHEA surrénalienne) en quantité suffisante fera pen-
cher la balance en faveur de labstention thérapeutique.
2.5. Les hyperandrogénies
Les syndromes dhyperandrogénie représentent lindica-
tion majeure du dosage des androgènes chez la femme.
Le syndrome des ovaires polykystiques saccompagne
fréquemment dune production anormale dandrogènes
avec élévation de la ∆4-androstènedione et, de façon plus
modeste et inconstante, de la testostérone sérique. Le
dosage de la testostérone libre ou de la testostérone
biodisponible, préconisé par certains auteurs (7), ne fait
pas lunanimité.
Dans le cadre des explorations, il faut garder à lesprit la
possibilité de tumeur virilisante de lovaire qui doit être

B. ARGEMI
228 Revue de l'ACOMEN, 1998, vol.4, n°3
soupçonnée et recherchée dès que le taux de testostérone
sérique dépasse 2 ng/ml (8).
Au cours des hirsutismes dits idiopathiques, où les an-
drogènes ovariens sont normaux, cest un trouble du mé-
tabolisme périphérique de la testostérone ou une hyper-
sensibilité des récepteurs qui est le plus souvent en cause.
On peut observer une élévation de la dihydrotestostérone
(DHT) ou de son métabolite landrostanediol dont le
glycuronide est plus facilement accessible aux méthodes
de dosage radioimmunologique (9).
Il ne faut pas oublier, enfin, que certains hirsutismes, avec
ou sans troubles de la fécondité, peuvent être en relation
avec une hyperplasie congénitale des surrénales par défi-
cit partiel en 21-hydroxylase. Lorsque lensemble du bilan
ovarien est négatif, un dosage de 17-hydroxy-progesté-
rone avec stimulation par lACTH doit être réalisé pour
léliminer avant de considérer quil sagit dun hirsutisme
idiopathique.
3. Exploration de la fonction testiculaire
Comme chez la femme, cest tout au long de la vie que lon
peut être confronté à des anomalies de la fonction
gonadique chez lhomme.
3.1. La cryptorchidie
Les chirurgiens ont tendance à opérer de plus en plus tôt
les testicules cryptorchides pour préserver la fertilité ulté-
rieure, mais il faut sêtre assuré auparavant de la présence
de testicules fonctionnels intra abdominaux. Celle-ci est
confirmée par laugmentation de la testostérone sérique
au-dessus de 3 ng/ml, après 7 injections de 1500 UI de
gonadotrophine chorionique administrées à 2 jours din-
tervalle.
3.2. Troubles de la puberté
3.2.1. Les pubertés précoces
A côté de la puberté précoce vraie par tumeur hypothala-
mique, plus rarement testiculaire, ou, le plus souvent idio-
pathique, il faut distinguer les pseudo-pubertés précoces
par hyperplasie congénitale des surrénales, et plus rare-
ment, la testotoxicose, par anomalie des protéines G au
niveau de la membrane des cellules de Leydig (10) et qui
sobserve habituellement chez des enfants plus jeunes.
Le Tableau I ci-dessous résume les caractères cliniques et
biologiques qui permettent de différencier ces trois condi-
- TABLEAU I -
Pseudo-puberté précoce Puberté précoce vraie Testotoxicose
Pilosité
Verge
Testicules
Augmentée
Augmentée
Normaux
Augmentée
Augmentée
Augmentés
Augmentée
Augmentée
Augmentés
LH
FSH
Testostérone
Basse
Basse
Basse
Augmentée
Augmentée
Augmentée
Basse
Basse
Augmentée
3.2.2. Les impubérismes
Labsence de tout signe de puberté après 15 ans doit faire
éliminer avant tout les différentes causes dhypogona-
disme : une dysgénésie gonadique (syndrome de
Klinefelter) est facilement rattachée à sa cause par la clini-
que et le caryotype ; le diagnostic biologique dun hypo-
gonadisme hypogonadotrophique repose sur les résultats
du dosage de FSH et LH après stimulation.
En labsence de cause organique, on peut alors parler de
retard pubertaire. Dans tous ces cas, le dosage de la tes-
tostérone sérique permet dévaluer la capacité fonction-
nelle du testicule endocrine. Le dosage de DHEA ou S-
DHEA en labsence de tout signe de puberté donne, comme
chez la fille, des informations sur le degré de maturité des
surrénales.
3.3. Lhomme adulte
3.3.1. Les hypogonadismes sont la plupart du temps se-
condaires. Le dosage de la testostérone entre dans le ca-
dre de lexploration globale du syndrome et indique le ni-
veau datteinte gonadique. Quil sagisse de la testosté-
rone totale, libre ou biodisponible, aucune de ces trois
méthodes de mesure na montré sa supériorité en dehors
de perturbations importantes de la SHBG.
3.3.2. Les troubles de la fertilité ne saccompagnent quex-
ceptionnellement de perturbations de la production de tes-
tostérone : les causes endocriniennes dhypofertilité sont
rares et habituellement évidentes.
3.3.3. Les dysérections dorigine endocrinienne chez
ladulte jeune sintègrent dans le cadre dun hypoandrisme
facilement rattaché à sa cause. Après la cinquantaine, le

Les indications du dosage des hormones stéroïdes
229
Revue de l'ACOMEN, 1998, vol.4, n°3
tions pathologiques.
dosage de testostérone, associé à celui de la FSH, permet
de faire le diagnostic de la peu fréquente andropause. Son
dépistage systématique à cette période de la vie est impor-
tant car le traitement substitutif donne alors dexcellents
résultats.
3.3.4. Les gynécomasties
En dehors de toute cause thérapeutique, on peut les ob-
server dans diverses circonstances pathologiques. Outre
la testostéronémie et le dosage de la prolactine, la mesure
de lestradiol sérique est importante pour dépister une tu-
meur féminisante du testicule ou de la surrénale.
Les cirrhoses hépatiques saccompagnent dun déséquili-
bre estrogènes/testostérone lié dune part à la baisse de
synthèse de la SHBG, dautre part aux perturbations du
métabolisme des stéroïdes et en particulier des estrogènes.
4. La cortico-surrénale
4.1. Rappel physiologique
La biosynthèse du cortisol par la zone fasciculée et celle
des androgènes dont le siège est principalement la zone
réticulée de la cortico-surrénale, sont sous la dépendance
de lACTH. Cest la concentration en cortisol qui assure le
rétro-contrôle négatif de la production hypothalamique de
C.R.H (corticotropin releasing hormone). La production
dACTH et de cortisol suit un rythme nycthéméral de
grande amplitude, avec une sécrétion maximale entre 4h et
8h du matin et un minimum entre 20h et 24h. La sécrétion
daldostérone par contre, au niveau de la zone glomérulée,
est indépendante de lACTH et soumise au contrôle du
système rénine-angiotensine.
Dans certaines conditions pathologiques, cette dualité peut
disparaître et être à lorigine, comme nous le verrons, dhy-
pertensions artérielles assez particulières.
4.2. Les hypercorticismes
4.2.1. Le syndrome clinique (Cushing) est commun à tous
les hypercorticismes. Il associe, dans sa forme complète,
une obésité facio-tronculaire avec «bosse de bison» cer-
vicale basse, des vergetures pourpres, une amyotrophie,
une hypertension artérielle, un diabète et enfin une démi-
néralisation osseuse.
4.2.2. Le syndrome biologique est celui dun hypercortiso-
lisme : augmentation du cortisol sérique, perte du rythme
nycthéméral, et élévation de lexcrétion urinaire du corti-
sol.
4.2.3. Le diagnostic étiologique repose dune part sur
limagerie, dautre part, sur les dosages dACTH et les
épreuves dynamiques (Tableau II).
Il est rendu parfois difficile par le caractère incomplet ou
- TABLEAU II -
Origine du Cushing
Test Tolérance Réponse normale
Hypothalamus Hypophyse Surrénales Para
néoplasique
Lysine-
vasopressine
CRH
Mauvaise
Bonne
ACTH
x 2 à 4 + + Sans intérêt ±
Hypoglycémie
inulinique
Métopirone
Correcte
Médiocre
ACTH
x 2 à 6 - + Sans intérêt -
Dexaméthasone
"Minute"
Faible
Fort
Bonne ACTH freinée
de plus de 50%
Cortisol sérique <10nM
Cortisol urinaire abaissé
++ - -
4.3. Les hypocorticismes
Quils soient primitifs (maladie dAddison) ou secondai-
res à un déficit hypophysaire, leur diagnostic biologique
ne pose pas de problème majeur.
4.4. Cas particulier de la corticothérapie au
long cours
Certaines affections rhumatismales chroniques, les
connectivites, les greffes dorganes ou lasthme sévère,
nécessitent une corticothérapie qui peut durer plusieurs
 6
7
6
7
1
/
7
100%