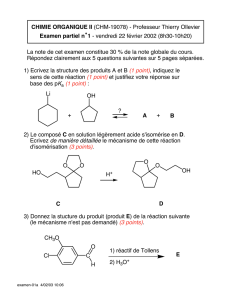
Chimie Organique: Phénols et Éthers - Cours Universitaire

Université Nangui Abrogoua Cours de Chimie Organique Prof. Békro Yves-Alain
UFR – SFA Licence 3 Prof. Mamyrbékova Békro Janat
1
Chapitre IX. PHENOLS
Chapitre IX. PHENOLSChapitre IX. PHENOLS
Chapitre IX. PHENOLS
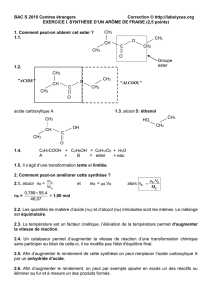
Les phénols sont des composés aromatiques contenant OH dans le cycle
benzénique.
OH OH OH OH OH
CH
3
phénol crésols
OH
OH OH
pyrogallol résorcine hydroquinone
OH
OH
HO
phloroglucine
1. Méthodes d’obtention
10% des phénols et des crésols sont obtenus à partir du goudron ; les 90% sont
des produits de synthèse.
1.1. Réaction alcaline (SN
ar
)
H
2
SO
4
SO
3
H
NaOH
HOH
SO
3
Na
NaOH
solide
350-450°C
- Na
2
SO
3
ONa
t°C
OH
H
+
2H
2
SO
4
SO
3
H
NaOH
HOH
SO
3
Na
2NaOH
solide
350-450°C
- 2Na
2
SO
3
ONa
t°C
SO
3
SO
3
HSO
3
Na
OH
H
+
OH OH
1.2. A partir des halogénures d’alkyles
6-8% NaOH
p, t°C
Cl
GED
OH
GED
NaOH
HOH t°C
Cl OH
GEA GEA
GED- groupement électrodonneur
(GEA) (GEA)
,
(SN
arome
)
(SN
arine
)
GEA - groupenent électroaccepteur
1.3. A partir d’anilines
HCl
NH
2
,
NaNO
2
0 - 5°C
NNCl
HOH, H
+
, t°
SN1
OH
+ H
2

Université Nangui Abrogoua Cours de Chimie Organique Prof. Békro Yves-Alain
UFR – SFA Licence 3 Prof. Mamyrbékova Békro Janat
2
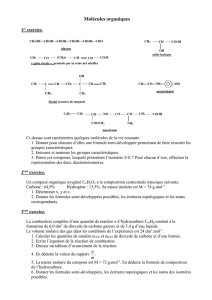
1.4. Méthode industrielle : procédé au cumène
Ce procédé a été mis au point en 1944 par Hock et Lang.
CH - CH
3
CH
3
O
2
100°C CH
3
CH
3
25°C
H
2
SO
4
+CH
3
- C - CH
3
=
O
C-O-OH
OH
Hydroperoxyde de cumène
1.5. Peroxydation catalytique du benzène
OH
HOOH, t°C
cat: Fe
3+
, Cu
2+
, Ti
4+
+ H
2
O
2. Propriétés chimiques
O - H H acide, propriété acide
liaison polaire, propriété nucléophile
2.1. Propriétés acides
C
2
H
5
OH
C
6
H
5
OH H
2
CO
3
CH
3
COOH
K
a
10
-
18
1,1x 10
-
10
2,7x 10
-
7
1,7x 10
-
5
pK
a
18 9,98 pK
a1
= 6,5
pK
a2
=10,3 4,76
Le phénol (encore appelé hydroxybenzène, acide phénique ou acide
carbolique) est plus acide que les alcools. L’anion phénolate (base conjugué du
phénol) est stabilisé par mésomérie.
C
2
H
5
OH + NaOH C
2
H
5
O
-
Na
+
+ H
2
O
Impossible d’obtenir l’alccolate
O - H
+ NaOH
O
+ H
3
O
Na

Université Nangui Abrogoua Cours de Chimie Organique Prof. Békro Yves-Alain
UFR – SFA Licence 3 Prof. Mamyrbékova Békro Janat
3
O O OO
O - MgBr
+CH
4
CH
3
MgBr
O - H
O - H
Na
2
CO
3
O - Na
CO
2
+ H
2
O
O - H
+ NaHCO
3
pas de réaction
• Les GED dans le cycle diminuent l’acidité des phénols :
O - H O - H O - H O - H O - H
Cl
NO2CH3OCH3
>>>>
pKa9,14 9,28 9,93 10,19 10,80
• Réaction d’identification des phénols :
O - FeCl
2
O - H
FeCl
3
à 5%
- HCl
précipité bleu-noire
2.2. Propriétés nucléophiles
Les phénols sont moins réactifs comme Nu
-
que les alcools.
O - H HCl
PCl
5
POCl
3
pas de réaction
1)

Université Nangui Abrogoua Cours de Chimie Organique Prof. Békro Yves-Alain
UFR – SFA Licence 3 Prof. Mamyrbékova Békro Janat
4
O - H
+ NaOH
ONa CH
3
I
-NaI
(CH
3
O)
2
SO
2
- Na(CH
3
)SO
4
O - CH
3
O - CH
3
2)
ONa
CH
3
CH
2
Br
-NaBr
O - CH
2
CH
3
3) (SN)
O - C = O
O - H
4) O-acylation
(CH
3
CO)
2
O
AN
CH
3
COONa
CH
3
COCl
pyridine
CH
3
O - C = O
(CH
3
CO)
2
OCH
3
COOH
CH
3
OH
COOH
acide salicilique
t°C COOH
aspirine
+
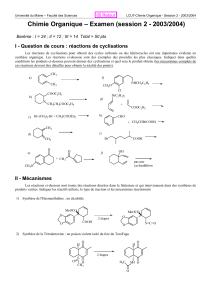
2.3. Réactions de SE
arom
OH
OH
CO
2
p, t°C
OH
COOH
réaction de
Kolbe-Schmitt
N = N
HO - réaction
d'azocondensation
HNO
3 dilué
NO
2
+
OH
NO
2
Br
2
CH
3
COOH
OH
Br
+
OH
Br
SE
arom
C
6
H
5
N
2
Cl
OH est un groupement activant (électro donneur) du cycle benzénique par + M
(–I et +M, M>I), par conséquent, le phénol est plus actif dans SE
arom
que le

Université Nangui Abrogoua Cours de Chimie Organique Prof. Békro Yves-Alain
UFR – SFA Licence 3 Prof. Mamyrbékova Békro Janat
5
benzène. Les premiers potentiels d’ionisation l’illustrent bien
(I
1
benzène = 9,26 eV, I
1
phénol = 8,5eV).
Sous contrôle orbitaire la première attaque s’effectue en position p- et la
seconde en o- sous contrôle de charge
.
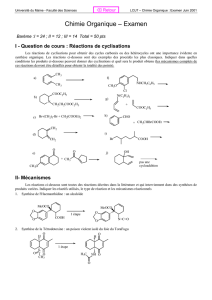
2.3.1. Réaction de Kolbe-Schmitt
OH
CO
2
p, 125°C
ONa
COOH
NaOH
ONa
HCl
OH
COOH
HOH
OH
CO
2
p, 250°C
OK
COOH
KOH
OK
HCl
OH
HOH
COOH
OOH
COOH
O
Mécanisme:
CO
O
O
H
CO
O
OH
CO
O
De la même manière, la réaction de formylation se réalise.
OH
H - C
=
H
O
H
+
OH CH
2
OH
+
OH
CH
2
OH
12
H - C
=
H
OH
+
H - C
=
H
OH
H - C
H
OH
L’alcool o-hydroxybenzylique (1) est utilisé pour l’obtention d’un polymère
important- la bakélite. On l'utilise pour ses propriétés isolantes et
thermorésistantes pour fabriquer les boîtiers de radios et de téléphones ; comme
isolant électrique dans les prises, interrupteurs et gaines de fils électriques, de
même que dans divers ustensiles de cuisine, de bijoux et de jouets.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
1
/
93
100%