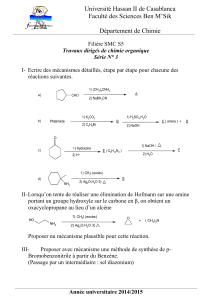
Révision Stratégie de synthèse - Anne CURK

REVISIONS (ET COMPLEMENTS) DE SYNTHESE ORGANIQUE 2016 2017........2
I- MODIFICATION DES CHAINES CARBONEES.......................................................................2
1- Création de liaison C-C par action d’un C nucléophile sur un C électrophile 2
1-
A
D
EFINITION DES ORGANOMAGNESIENS
2
1-
B
P
ROPRIETES DES ORGANOMAGNESIENS
2
1-
C
S
TRUCTURE DES ORGANOMAGNESIENS
2
1-
D
- R
EACTIVITE DES ORGANOMAGNESIENS
(N
OUVEAUTES
)
2
Sur les aldéhydes et cétones, CO
2
, époxydes et esters.
2- Rupture de liaison CC : d’un diol
α
HO-C-C-OH ou d’un alcène C=C 6
2-
A DE L
’
ALCENE AU DIOL
α :
OXYDATION CATALYTIQUE PAR
O
S
O
4
ET
NMO 6
2-
B
C
LIVAGES OXYDANTS DE DIOLS
α
PAR
N
A
IO
4
7
2-
C
C
LIVAGE DIRECT DE L
’
ALCENE PAR
O
S
O
4
ET
N
A
IO
4
(
DIOL
α
INTERMEDIAIRE NON ISOLE
) 7
II-CONVERSION DE GROUPES CARACTERISTIQUES............................................................8
Présentation des groupes caractéristiques en classes d’oxydation 8
1- Conversions par oxydoréduction
(AVEC NOUVEAUTES)
8
1-
A
L
ES OXYDATIONS
8
Oxydation des alcènes diol α 8
Oxydation des alcènes époxydes
(Nouveauté)
8
Oxydation des alcools en aldéhyde, cétone ou acide carboxylique 9
Oxydation des aldéhydes en acides carboxyliques 10
1-
B
L
ES REDUCTIONS
10
Obtention des organomagnésiens : de R-X à R-Mg-X. 10
Les réactions parasites 11
Réduction des aldéhydes et cétones en alcools 12
Réduction des esters en alcools primaires, par LiAlH
4(NOUVEAUTE)
13
Le DIBAL-H : réduction ménagée des esters en aldéhydes
(NOUVEAUTE)
13
Réduction des acides carboxyliques
(NOUVEAUTE)
15
2- Conversion au sein d’un même groupe rédox 17
2-
A
S
UBSTITUTIONS NUCLEOPHILES SUR LES
R-X
ALCOOLS
,
ETHERS
,
EPOXYDE
17
2-
B
E
LIMINATIONS SUR
R-X
ET
A
LCOOLS
ALCENES
17
2-
C
A
DDITIONS NUCLEOPHILES D
’
ALCOOLS SUR
C=O =>
HEMIACETALS ET ACETALS
18
3- Protection de fonctions 20
3-
A
P
ROTECTION DU GROUPE ALCOOL
20
Obtention de l’acétal de la DHP 20
Obtention d’éthers par Williamson 21
Obtention d’éther silylé 22
3-
B
P
ROTECTION DU GROUPE
C=O
DES ALDEHYDES ET CETONES
23
3-
C
P
ROTECTION DES DIOLS
α 23
4- Activation de fonctions 23
4-
A
A
CTIVATION NUCLEOPHILE DES ALCOOLS
: R-OH
R-O
-
24
4-
B
A
CTIVATION ELECTROPHILE DES ALCOOLS
MEILLEUR GPE PARTANT
24
4-
C
A
CTIVATION ELECTROPHILE DE
C=O
DES ALDEHYDES ET CETONES PAR LES ACIDES
24
FICHE : OBTENTION DES EPOXYDES
( AVEC 1 NOUVEAUTE)
.......................................27
I - A PARTIR D'UN ALCOOL α HALOGENE OU α HALOGENO ALCOOL (REVISION) ..........27
II - A PARTIR D'UN ALCENE
(NOUVEAUTE)
....................................................................................28

2
REVISIONS (ET COMPLEMENTS) DE SYNTHESE ORGANIQUE 2016 2017
Votre cours de chimie organique est orienté pour vous permettre de créer ( ou d’analyser ) des stratégies de synthèse
organique. Ainsi, il est construit en 2 grands chapitres : modification des chaînes carbonées, et conversion des
groupes caractéristiques.
I- MODIFICATION DES CHAINES CARBONEES
Il s’agit ici, de créer des liaison C-C, avec allongement de chaîne, ou de construire des cycles, ou encore de couper
des liaisons C-C.
1- Création de liaison C-C par action d’un C nucléophile sur un C électrophile
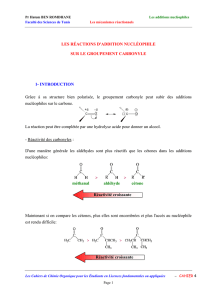
Les organomagnésiens RMgX sont porteurs d’un carbone nucléophile, et synthétisés dans ce seul but. Ils sont un cas
particulier parmi des organometalliques
1-
A
D
EFINITION DES ORGANOMAGNESIENS
On appelle organométallique tout composé présentant une liaison C – Métal
Exemples : CH
3
– CH
2
– Mg – Br organomagnésien
CH
3
Cu , Li
CH
3
organocuprate lithié
1-
B
P
ROPRIETES DES ORGANOMAGNESIENS
Ils sont tous fabriqués dans un seul but : LE METAL DONNE AU CARBONE LIE UNE POLARITE
δ
-
Le doublet de la liaison C Métal est alors à la fois BASIQUE et NUCLEOPHILE
δ
-
δ
+
Par exemple CH
3
– CH
2
– MgBr / CH
3
– CH
2
– H
Base acide ( l'acide est un alcane : pK
A
≈ 40 )
Pour tous , les acides conjugués étant des alcanes ( ou alcènes , ou alcynes ) , ce sont des BASES TRES FORTES.
Toutefois, en absence de tout proton acide, ce doublet C – Métal a des PROPIETES NUCLEOPHILES TRES
MARQUEES.
1-
C
S
TRUCTURE DES ORGANOMAGNESIENS
R Mg R Le magnésium présente deux lacunes électroniques .
Dans cet état il est instable et impossible à obtenir.
Il n'existe que stabilisé par solvatation, en présence de DOUBLETS D'ELECTRONS appartenant au solvant :
R Mg X
S
S
où S est un solvant, INDISPENSABLE , donneur de doublet
Ainsi stabilisé, le magnésium est tétravalent (règle de l'octet respectée )
1-
D
- R
EACTIVITE DES ORGANOMAGNESIENS
(N
OUVEAUTES
)
Les organomagnésiens permettent d'allonger les chaînes carbonées, par création de liaison C – C en réagissant, par
addition nucléophile, sur les
CH
3
– Zn – Br organozincique
Pb-(-CH
2
– CH
3
)
4
Plomb tétra éthyl
CH
3
– Cd – CH
3
Organocadmien

3
♦Aldéhydes et cétones
O
R
1
R
2
δ
+
+
RMgX
R
1
R
2
O
-
R
MgX
+R
1
R
2
OH
R
+
Mg
2+
+
1° étape, milieu anhydre
addition nucléphile
2° étape , hydrolyse acide
H
2
O , H
+
X
-
éther anhydre A/B
(sel)
On a donc créé une liaison C-C au niveau du C électrophile du groupe carbonyle C=O, groupe qui a disparu, se
transformant en alcool, au minimum secondaire.
♦CO
2
O C O
δ
+
+
R MgX
addition
nucléophile
éther anhydre
RO
O
-
MgX
+
RO
OH
+
Mg
2+
+
X
-
H
2
O , H
+
A/B
(sel) acide carboxylique
δ
−
On remarquera que RMgX n'agit donc que sur des composés ELECTROPHILES et INSATURES ( liaison π ou cycle ),
par addition nucléophile.
O C O
δ
+
+
R MgX
addition
nucléophile
éther anhydre
RO
O
-
MgX
+
RO
OH
+
Mg
2+
+
X
-
H
2
O , H
+
A/B
(sel) acide carboxylique
δ
−
♦
♦Epoxydes
(Nouveauté)
Voir la fiche "obtention des époxydes" proposée en fin de ce chapitre (au programme )
(Nouveauté)
.
Les époxydes sont des composés très réactifs, et en particulier sensibles aux nucléophiles. En effet ils contiennent un
carbone électrophile ( donc action d'un nucléophile possible ) et insaturé ( par le cycle, donc addition possible )
O
H
δ
+
δ
+
R MgX
addition
nucléophile
OH
R
Mg X
site le moins encombré : REGIOSELECTIVE
attaque anti : STEREOSPECIFIQUE
OH H
R
H
2
O , H
+
A/B
(sel)
+
Mg2+
+
X-
1° étape en solvant anhydre, éther ou THF
2°étape : hydrolyse acide pour libérer un alcool
On a donc allongé la chaîne carbonée :

4
• Si l'on regarde l'époxyde : on a ajouté un groupe R sur le site le moins encombré de l'époxyde, une fonction
alcool se trouvant sur le C en α.
• Si l'on regarde le dérivé halogéné que l'on avait transformé en organomagnésien : on a rajouté au minimum
2 carbones : ceux porteurs de la fonction époxyde. La fonction alcool qui est apparue est en β par rapport au
C initialement porteur du dérivé halogéné.
♦Esters
(Nouveauté)
La fonction ester réagit sur les organomagnésiens car elle est :
• Porteuse d'un carbone électrophile ( donc sensible à un nucléophile ) ET insaturé ( par la double liaison =O
, et donc addition possible )
Mais elle réagit différemment des aldéhydes et cétones car elle est :
• Porteuse en plus du groupe attracteur d'électron O – R
2
sur le C=O, se comportant comme un groupe
partant.
R1O
OR2
Ph
O
O
+
R MgX
Ph
O
-
R
O
MgX
Ph
R
O
+
2° action , INEVITABLE
R MgX
H
2
O , H
+
Ph
R
R
OH
+
Mg
2+
+
X
-
δ
+
δ
+
δ
−
add Nu
éther anhydre
Décomposition
spontanée
INEVITABLE
(sel instable)
(sel)
A/B
Ph
O
-
R
R
MgX
fin de la 1° étape, en solvant base de Lewis anhydre
2° étape : hydrolyse acide
+
+
groupe
acyle
groupe partant
alcoolate
et simultanément
H
2
O , H
+
A/B
+ Mg
2+
+ X
-
O
-
MgX
+
O
-
MgX
+
OH
ESTER : δ
+
Nomenclature des esters : Alcanoate d'alkyl
Propanoate de phényle
Benzoate de méthyle
2-chloro éthanoate de 3-méthyl benzyle
L'organomagnésien se fixe
• Une première fois par addition Nucléophile suivie d'élimination. AN/E
• Une deuxième fois par addition nucléophile sur l'aldéhyde ou cétone intermédiaire non isolé : AN
On obtient donc un alcool tertiaire, avec deux groupes R identiques, provenant de 2 molécules d'organomagnésien
fixées.
♥ Un organomagnésien se fixe 2 fois sur un ester.

5
Exemples concrets d'application :
Proposer une méthode de synthèse du triphénylcarbinol ( ou tri phényl méthanol ) à partir de bromo
benzène comme seul produit organique. Pourquoi observe-t-on une couleur rouge lorsqu'on place ce
produit en milieu acide?
Présenter le mécanisme d'action du chlorure de méthylmagnésium sur la lactone (a) ci-contre :
( Une lactone est un ester cyclique ) . En déduire une méthode d'obtention de l'éther cyclique (b).
O
O
O
(a)
(b)
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
1
/
31
100%