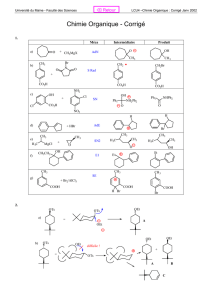
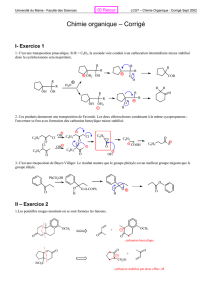
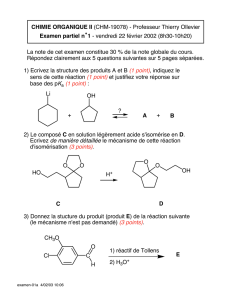
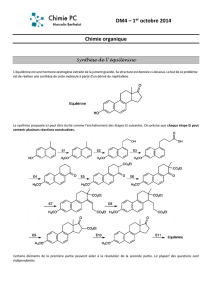
2004

Stéphane Cosandey
TP de chimie organique 3ème année
Décembre 2004
Assistante : Ségolène Gille
2-Méthylcyclopentanone

2
Introduction
A partir de l’acide adipique on veut synthétiser le 2-méthylcyclopentanone en 4 étapes ;
estérification, cyclisation, méthylation et décarboxylation.
CO2H
CO2HEtOH/H
Toluene CO2Et
CO2Et Na
O
CO2Et
O
O
CO2Et
NaH
CH3I
Toluene
H
Cette molécule peut voir des applications dans les produits servant à faire fuir les insectes.
Mécanisme
La première étape consiste en une estérification de l’acide adipique. Dans un premier temps
l’acide carboxylique est protoné par l’acide sulfurique concentré, ensuite l’oxygène de
l’éthanol attaquera le carbone électrophile. On assistera enfin au départ d’une molécule
d’eau :
O
OH
HO
O
H
O
OH
HO
O
H
O
OH
HO
O
CH3CH2OH
H
OH
H3CH2C
- H
O
OH
HO
O
H
O
H3CH2C
H
O
O
HO
O
H
O
H3CH2C
H
H
- H2O; H
O
HO
O
OEt

3
Le processus se répète pour la deuxième fonction alcool de la molécule afin d’obtenir le
diéthyl adipate.
La deuxième étape est une cyclisation intramoléculaire (réaction de Claisen
intramoléculaire, ou condensation de Dieckmann). Cependant le départ de la réaction est un
peu différent d’une condensation de Dieckmann conventionnelle. En effet on traite la
molécule directement avec le sodium (sans passer par un éthanoate) obtenant ainsi un radical
qui va cycliser et donner dans le même temps deux équivalent éthanoate. Cet alors ces
éthanoates qui déprotonerons les hydrogènes en alpha de l’ester des molécules restantes. On
aura alors un carbanion qui attaquera le carbone électrophile de la deuxième fonction ester de
la molécule, obtenant ainsi la molécule cyclique (après un départ d’éthanoate qui explique la
quantité catalitique nécessaire) :
O
EtO
O
OEt
+2Na
CO
2
Et
CO
2
Et
Na
Na
O
OEt
OEt
O
O
O
+ 2 EtONa
...
La réaction peut ensuite encore s’arranger selon la présence de sodium dans le milieu, mais
ceci nous intéresse moins que la formation de l’éthanoate qui va servir pour la suite :
O
EtO
O
OEt Na
+
HH
O
EtO
O
OEt
O
EtO
O
OEt
O
OEt
O
OEt
O
OEt
O
EtOH +
HCl
O
OEt
O
OEt
Ce proccesus, bien que faisant perdre un peu de produit par une cyclisation secondaire, est
cependant plus efficace que la démarche conventionelle. Cela peut s’expliquer en terme de
thermodynamique par la différence de pKa. En effet le carbanion obtenu avant acidification
étant le plus stable la réaction est favorisée. Cependant l’éthanol ayant un pKa proche (~16

4
pour l’éthanol et 14 pour l’ester) la réaction est facilement réversible. A l’inverse dans le
toluène comme solvant (pKa >30) la réaction est plus volontiers stabiliser vers les produits.
On assiste ensuite à une méthylation :
O
H
OEt
O
NaH
O
OEt
O
CH
3
I
O
OEt
O
CH
3
+NaI
- H
2
Enfin on procède à une décarboxylation après acidification :
O
CH
3
O
OEt H
O
CH
3
O
OEt
HO
CH
3
O
OEt
O
CH
3
OH
OEt
H
H
O
H
O
CH
3
HO OEt
O
H
H
- H
O
CH
3
HO OEt
OH
H
O
CH
3
HO OEt
OH
H
O
CH
3
O
OH
H
+ EtOH
O
CH
3
O
OH
HO
CH
3
O
OH
O
CH
3
O
OH
HH
O
CH
3
O
OH
Cette réaction à lieu dans ce sens grâce à l’excès d’eau par rapport à la formation d’éthanol.
O
CH3
O
OH
O
CH3
O
O
H
OH
CH3+ CO2
O

5
Mode opératoire
Etape 1
CO
2
H
CO
2
HEtOH/H
Toluene CO
2
Et
CO
2
Et
diethyl adipate (48.75 g ; 96%)
acide
adipique éthanol toluène a.
sulfurique
MM [g/mol] 146.1 46.07 92.14 98.08
densité
[g/ml] 0.81 0.87 1.83
moles 0.25 1.6 0.42 3.8*10-3
grammes 36.5 0.37
ml 90 45 0.2
On place de l’acide adipique (36.5 g, 0.25 mol), de l’éthanol (90 ml, 1.6 mol), du toluène
(45 ml) et de l’acide sulfurique concentré (0.2 ml, 3.8 mmol) dans un ballon de 250 ml. Le
ballon est rattaché à une colonne vigreux surmonté d’une tête de distillation. Le tout est
chauffé à 115°C. Une fois la dissolution de l’acide adipique complète on observe une
distillation d’un mélange azéotropique d’alcool de toluène et d’eau (autour des 75°C). le
mélange est récupéré dans un ballon contenant 38 grammes de carbonate de potassium
anhydre (ce afin de piéger l’eau). La distillation est poursuivie jusqu’à l’augmentation de la
température au sommet de la colonne à 78°C. La solution récupérée dans le ballon est filtrée
puis réintroduite dans le mélange de départ. Le tout est alors distillé à pression réduite, où
l’alcool et le toluène passe rapidement, et on récupère notre produit, le diéthyl adipate (32.4
g ; 64%)à 142°C (pour 28 mmHg).
Etape 2
CO
2
Et
CO
2
Et Na
O
CO
2
Et
Toluene
11.6 g ; 74%
 6
7
8
9
6
7
8
9
1
/
9
100%