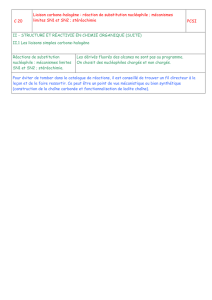
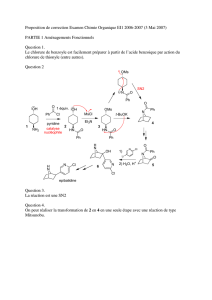
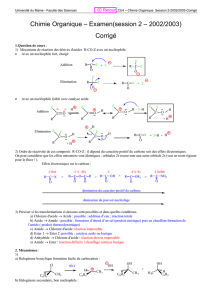
Les Substitutions Nucléophiles Sur les carbones saturés

L
LL
L
L
LL
L
L
LL
Le
ee
e
e
ee
e
e
ee
es
ss
s
s
ss
s
s
ss
s
S
SS
S
S
SS
S
S
SS
Su
uu
u
u
uu
u
u
uu
ub
bb
b
b
bb
b
b
bb
bs
ss
s
s
ss
s
s
ss
st
tt
t
t
tt
t
t
tt
ti
ii
i
i
ii
i
i
ii
it
tt
t
t
tt
t
t
tt
tu
uu
u
u
uu
u
u
uu
ut
tt
t
t
tt
t
t
tt
ti
ii
i
i
ii
i
i
ii
io
oo
o
o
oo
o
o
oo
on
nn
n
n
nn
n
n
nn
ns
ss
s
s
ss
s
s
ss
s
N
NN
N
N
NN
N
N
NN
Nu
uu
u
u
uu
u
u
uu
uc
cc
c
c
cc
c
c
cc
cl
ll
l
l
ll
l
l
ll
lé
éé
é
é
éé
é
é
éé
éo
oo
o
o
oo
o
o
oo
op
pp
p
p
pp
p
p
pp
ph
hh
h
h
hh
h
h
hh
hi
ii
i
i
ii
i
i
ii
il
ll
l
l
ll
l
l
ll
le
ee
e
e
ee
e
e
ee
es
ss
s
s
ss
s
s
ss
s
S
SS
S
S
SS
S
S
SS
Su
uu
u
u
uu
u
u
uu
ur
rr
r
r
rr
r
r
rr
r
l
ll
l
l
ll
l
l
ll
le
ee
e
e
ee
e
e
ee
es
ss
s
s
ss
s
s
ss
s
c
cc
c
c
cc
c
c
cc
ca
aa
a
a
aa
a
a
aa
ar
rr
r
r
rr
r
r
rr
rb
bb
b
b
bb
b
b
bb
bo
oo
o
o
oo
o
o
oo
on
nn
n
n
nn
n
n
nn
ne
ee
e
e
ee
e
e
ee
es
ss
s
s
ss
s
s
ss
s
s
ss
s
s
ss
s
s
ss
sa
aa
a
a
aa
a
a
aa
at
tt
t
t
tt
t
t
tt
tu
uu
u
u
uu
u
u
uu
ur
rr
r
r
rr
r
r
rr
ré
éé
é
é
éé
é
é
éé
és
ss
s
s
ss
s
s
ss
s
I
I_
_
G
Gé
én
né
ér
ra
al
li
it
té
és
s
A
A.
.
I
In
nt
tr
ro
od
du
uc
ct
ti
io
on
n
La substitution nucléophile consiste à remplacer un groupe partant par un
nucléophile. Il existe deux manières différentes de procéder :
_ Le groupe partant se sépare de la molécule, qui forme alors un
carbocation. Celui-ci se fait ensuite attaquer par le nucléophile. On obtient un
mélange racémique. Cette réaction est une SN
1
.
_ Le nucléophile se lie à la molécule en chassant le groupe partant, en une
seule étape. On obtient l'énantiomère inverse. Cette réaction est une SN
2
.
B
B.
.
S
St
ta
ab
bi
il
li
it
té
é
d
du
u
c
ca
ar
rb
bo
oc
ca
at
ti
io
on
n
Un carbocation est un intermédiaire, où un carbone a une case électronique
vide. Plus un carbocation est substitué, plus il est stable.
Cette accroissement de stabilité est du à des effets d'hyperconjuguaison.
Lorsqu'un groupement CH
3
(entre autres) est lié au carbocation, les orbitales de
R
R''
R' X
Nu
-
R
X
R''R' R''
R'
R
Nu
-
Inversion de Walden
R
R'
X
R''
R
R'
Nu
-
R''
R
R'
Nu R''
+
Mélange racémique
Nu
-
R
R'
+
Cl
-
CH
3
H H
H
Est plus
stable que Est plus
stable que

chaque hydrogène se recouvrent plus ou moins avec l'orbitale vide du carbocation,
stabilisant ainsi sa charge positive.
Certains carbocations peu substitués peuvent tout de même être formés. Ils
sont alors stabilisés par mésomérie.
C
C.
.
N
No
ot
ti
io
on
n
d
de
e
g
gr
ro
ou
up
pe
e
p
pa
ar
rt
ta
an
nt
t
Pour qu'un groupement soit considéré un bon groupe partant, il doit satisfaire
certaines conditions :
L'anion formé est une base faible, il est donc faiblement lié au carbone
d'origine
La molécule formée est stable
Ex :
Le meilleur groupe partant est le triflate. C'est une base très faible (son acide
conjugué est très puissant ) et sa charge négative est très stabilisée par résonance
et grâce aux effets inductifs forts de —CF
3
. De plus les effets mésomères et les
liaisons fortement covalentes (atomes très électronégatifs avec des atomes de
valence élevée) maintiennent fortement la stabilité de ce groupement.
Un groupement peut être activé pour devenir un bon groupe partant. Pour
cela un autre atome peut se lier ou son nuage électronique peut être modifié, ou bien
une réaction intramoléculaire le pousse à partir.
Ex :
Carbocation
benzylique Carbocation
allylique
N N N
2
Molécule
stable
Br Br
Base
faible
FN'est jamais
groupe partant
OS
O
O
CF
3
La charge négative est
répartie sur les oxygènes
par mésomérie.
Forte attraction inductive
de la part des fluors.
O
Mauvais groupe
partant
Cl
Bon groupe
partant
Cl
Départ déclenché
H
H
O
H
Excellent groupe
partant
Mauvais groupe
partant
AlCl
3
Cl
Al
Cl Cl Cl
HO

I
II
I_
_
S
SN
N
1
1
A
A.
.
G
Gé
én
né
ér
ra
al
li
it
té
és
s
Le mécanisme de la SN
1
s'effectue en deux étapes successives : le départ du
groupe partant, en formant un carbocation, puis l'attaque nucléophile sur la charge
positive.
La première étape est la plus lente, on dit
qu'elle est cinétiquement déterminante. La
vitesse de la réaction est donc influencée par la
facilité avec laquelle groupe partant se sépare de
la molécule, et par la stabilisation, ou solvatation,
du carbocation et de l'anion formé.
La vitesse de cette réaction ne dépend
alors que de la concentration de la molécule de
départ :
v = k
1
[RX]
.
B
B.
.
S
St
té
ér
ré
éo
oc
ch
hi
im
mi
ie
e
Le carbocation formé lors de la SN
1
est plane, son orbitale
p vide parallèle à ce plan. Le nucléophile peut donc attaquer d'un
côté ou de l'autre. On forme alors deux molécules
énantiomères.
Idéalement, on forme un mélange racémique ; cependant
ce cas est peu fréquent. La stéréochimie des composés dépend
de plusieurs facteurs.
l
l
I
In
nf
fl
lu
ue
en
nc
ce
e
d
de
e
l
la
a
d
di
is
ss
so
oc
ci
ia
at
ti
io
on
n
d
du
u
g
gr
ro
ou
up
pe
e
p
pa
ar
rt
ta
an
nt
t
Lorsqu'un groupe partant n'est pas facilement dissocié du carbocation,
l'attaque du nucléophile est influencée ; les énantiomères ne sont alors plus formés
de manière équimolaire.
La réaction tend alors plutôt vers un mécanisme de type SN
2
.
Ex :
C
C.
.
R
Ré
éa
ac
ct
ti
iv
vi
it
té
é
1
1)
)
I
In
nf
fl
lu
ue
en
nc
ce
e
d
de
e
l
la
a
s
st
tr
ru
uc
ct
tu
ur
re
e
E
t
R—X
R—Nu
R
+
Nu Nu
C
6
H
14
H
CH
3
Br
Et—OHC
6
H
14
HCH
3
Br
Nu
-
Plus lent
C
6
H
14
H
CH
3
+
C
6
H
14
HCH
3
83 % 17 %

Plus un carbocation est stable, plus il se forme facilement. Ainsi, de manière
générale, une molécule conduisant à un carbocation tertiaire sera plus réactive
qu'avec un carbocation secondaire ou primaire.
Cependant le reste de la structure de la molécule influence aussi sa réactivité.
On trouve ainsi plusieurs exceptions à la règle.
l
l
C
Ca
ar
rb
bo
oc
ca
at
ti
io
on
n
n
no
on
n
p
pl
la
an
ne
e
Lorsque le carbocation formé ne peut pas adopter une géométrie plane, à
cause des contraintes de la structure de la molécule, celui-ci ne se forme pas (ou
peu), car il n'est plus stabilisé par les effets d'hyperconjugaison.
l
l
D
Dé
éc
co
om
mp
pr
re
es
ss
si
io
on
n
s
st
té
ér
ri
iq
qu
ue
e
Dans une molécule encombrée, si le fait de former le carbocation diminue
grandement l'encombrement stérique, le départ du groupe partant est favorisé.
l
l
C
Ca
as
s
d
de
es
s
c
cy
yc
cl
le
es
s
Dans un cycle à six carbones, on observe un encombrement stérique dû aux
interactions 1,3-diaxiales. Si le départ du groupe partant diminue cet encombrement,
la formation du carbocation sera favorable.
2
2)
)
I
In
nf
fl
lu
ue
en
nc
ce
e
d
de
e
l
la
a
m
mé
és
so
om
mé
ér
ri
ie
e
Le départ d'un groupe partant peut être déclenché grâce à un effet mésomère.
Le carbocation formé se forme alors plus facilement, il est stabilisé par résonance.
l
l
H
Hé
ét
té
ér
ro
oa
at
to
om
me
e
a
av
ve
ec
c
u
un
n
d
do
ou
ub
bl
le
et
t
n
no
on
n-
-l
li
ia
an
nt
t
Le départ du groupe partant est activé grâce à un doublet non-liant.
X
Carbocation non plane
Défavorable bien qu'il
soit tertiaire
X
H
3
C
CH
3
CH
3
CH
3
CH
3
Néopentyle
H
3
C
CH
3
CH
3
CH
3
CH
3
Décompression stérique
Favorable bien qu'il soit
primaire
X
R
R
X
R
Assistance stérique
au départ du groupe
partant
Selon la
gène stérique de R
,
le départ du groupe partant est
favorisé
Favorable
OXO O

l
l
S
St
ta
ab
bi
il
li
is
sa
at
ti
io
on
n
p
pa
ar
r
m
mé
és
so
om
mé
ér
ri
ie
e
Le départ du groupe partant forme un carbocation stabilisé par mésomérie.
Cette réaction est favorable.
e
ee
e Le carbocation le moins substitué est le plus réactif.
Ex :
3
3)
)
E
Ex
xe
em
mp
pl
le
es
s
d
de
e
v
vi
it
te
es
ss
se
e
d
de
e
r
ré
éa
ac
ct
ti
io
on
n
d
de
e
S
SN
N
1
1
Molécule
Vitesse 0,07 0,12 2100 1 91 7700 130 000
Carbocation
D
D.
.
R
Ré
éa
ar
rr
ra
an
ng
ge
em
me
en
nt
ts
s
On observe un réarrangement lorsque celui-ci permet de former un
carbocation plus stable. Le réarrangement peut être une liaison entre deux Carbone
ou un déplacement d'un Hydrogène.
Ex :
X
X
Nu
Nu
-
Plus réactif Majoritaire
Cl
Cl
Cl
Cl
Cl
Primaire
Secondaire
Tertiaire
Primaire
Primaire
Cl
Primaire
Secondaire
Cl
Primaire
Secondaire
Délocalisé
Primaire
Tertiaire
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%