Intro

- 1 -
L’infection d’ascite
H Lécuyer, F Bert
Laboratoire de Microbiologie, Hôpital Beaujon (APHP)
1 Les infections chez le patient cirrhotique
La cirrhose est une maladie dont la prévalence est estimée en France entre 1500 et 2500
cas par million d’habitants et dont l’incidence est comprise entre 150 et 200 nouveaux cas par an
par million d’habitants. Seuls 100 000 malades ont une cirrhose décompensée et sont
symptomatiques 1. Cette maladie est responsable de 15 000 décès par an, les infections
représentant, avec 25% des décès, la deuxième cause de mortalité 2, 3.
Le cirrhotique est particulièrement sujet aux infections bactériennes du fait de sa
maladie : la cirrhose entraîne en effet une diminution des capacités phagocytaires du système
réticulo-endothélial (SRE, composé des cellules endothéliales et des cellules de Küppfer situées
au niveau des capillaires sinusoïdes hépatiques), liée sans doute à l’existence de shunt
intrahépatique, ou à l’altération des fonctions des macrophages 4, 5. Ainsi Hassner et al ont montré
que les monocytes présentent des capacités de phagocytose et de cytotoxicité diminuées 6. La
diminution de l’activité phagocytaire du SRE a été confirmée chez le malade, avec un
parallélisme entre la diminution de la captation de particules isotopiques par le SRE et le risque
d’infections du liquide d’ascite (ILA) ou de bactériémies 7, 8. Il existe également chez le
cirrhotique une diminution du complément sérique et donc des capacités d’opsonisation 9.
Runyon et al ont ainsi montré une relation inverse entre la diminution de l’activité opsonisante du
liquide d’ascite et le risque d’infection de celui-ci 10.
Les cytokines pro-inflammatoires sécrétées en réponse à l’infection participent à
l’altération progressive du tissu hépatique, au développement de l’encéphalopathie, et aux
troubles hémodynamiques qui aggravent l’hypertension portale et le syndrome hépato-rénal 11-13.
En dehors même de toute infection, des cytokines (TNF, IFN, IL6, IL12...) et des effecteurs
toxiques de l’inflammation (monoxyde d’azote, radicaux oxygénés...) sont sécrétés en réponse au
passage dans la circulation sanguine de composés bactériens (ADN, LPS …) provenant de la
flore digestive, réalisant une véritable « endotoxinémie intestinale » 14.
L’infection est également un facteur de risque important de récidive précoce
d’hémorragies digestives, ces hémorragies étant par ailleurs elles-mêmes facteurs de risque
d’infection 15, 16.
Ainsi, l’évolution de la maladie cirrhotique est un véritable cercle vicieux où la
détérioration progressive du tissu hépatique favorise les infections bactériennes, qui favorisent
elles-mêmes certaines complications de la cirrhose.

- 2 -
Les infections bactériennes représentent donc un événement majeur dans l’évolution
d’une cirrhose ; elles témoignent de la sévérité de la maladie hépatique, et sont un facteur
accélérant l’évolution de la maladie 17. Elles touchent près de 50% des malades hospitalisés. Les
ILA sont les plus fréquentes (31%), suivie des infections urinaires (25%) et des pneumopathies
(21%). Le taux de mortalité est élevé notamment pour les infections respiratoires (40%) et les
infections du liquide d’ascite (30%) 2.
2 Formation de l’ascite
L’ascite correspond à un épanchement liquidien dans la cavité péritonéale.
Cette accumulation de liquide peut être d’origine maligne (carcinose péritonéale) et
compliquer un cancer digestif ou ovarien ; elle peut également survenir au cours de la
tuberculose, de maladie pancréatique (rupture des canaux pancréatiques dans le péritoine) ou
d’insuffisance cardiaque.
Mais l’ascite survient la plupart du temps au décours de maladies hépatiques compliquées
d’hypertension portale et en particulier au cours de la cirrhose. L’hypertension portale est en effet
un facteur essentiel à la formation de l’ascite : l’élévation des pressions intra sinusoïdales entraîne
une fuite liquidienne vers la circulation lymphatique. Lorsque les capacités de drainage du canal
thoracique sont dépassées, cette fuite liquidienne se fait vers la cavité péritonéale. La formation
de ce troisième secteur entraîne une baisse de la volémie sanguine, qui conduit à l’activation des
systèmes sympathiques et rénine-angiotensine-aldostérone, et donc à une rétention rénale hydro-
sodée, augmentant l’hypertension portale. Parallèlement, la diminution de la production
hépatique des protéines plasmatiques entraîne une baisse de la pression oncotique, ce qui amplifie
le phénomène de fuite liquidienne.
3 Epidémiologie
L’ILA est une des complications les plus graves et les plus fréquentes chez le patient
cirrhotique. La prévalence de l’ILA chez les malades hospitalisés souffrant d'ascite est de 8 à
35% 2, 18, 19. Chez les malades asymptomatiques non hospitalisés, la prévalence de l'ILA est
faible, de l'ordre de 5%.

- 3 -
4 Physiopathologie : La translocation bactérienne
L’ILA consiste en une péritonite bactérienne primaire ou spontanée (« spontaneous
bacterial peritonitis » pour les Anglo-Saxons), où l’ascite est infectée à l’occasion d’une
bactériémie. Cette définition exclue les péritonites secondaires à une perforation digestive ou à
une fistule, avec passage direct des bactéries du tube digestif dans la cavité péritonéale.
En dehors des cas d’ILA consécutives à une septicémie à point de départ non digestif (par
exemple, lors de pyélonéphrites), la plupart des ILA sont la conséquence d’une translocation
bactérienne, c’est à dire du passage de bactéries du tube digestif dans les ganglions lymphatiques,
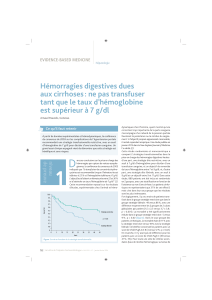
puis d’une bactériémie transitoire et d’un passage dans le liquide d’ascite (figure 1).
FIGURE 1.
Physiopathologie de l’infection du liquide d’ascite.
d’après Such et Runyon, Clin Infect Dis 1998 ;27 :669-76 20.
Hypertension portale
Pullulation
Translocation
bactérienne
Bactéries présentes dans les
ganglions mésentériques
Facteurs de virulence
bactériens ?
Altérations de l’épithélium
Altérations de l’immunité locale
Bactériémie
« Ensemencement » du
liquide d’ascite
Infection du
liquide d’ascite
Hypomobilité intestinale
Altérations de l’immunité
locale
Réponse inflammatoire
Bactérascite
Pas de réponse inflammatoire

- 4 -
La translocation bactérienne est un mécanisme complexe, conséquence de la rupture de
l’équilibre subtil qui existe dans le tube digestif entre colonisation bactérienne et défenses de
l’hôte.
Le tube digestif est en effet largement colonisé par des bactéries : on estime qu’il existe
105 unités-formant-colonies (ufc) par mL dans le jéjunum, 108 ufc/mL dans l’iléum et jusqu’à
1012 ufc/mL dans le colon 21. Cette flore digestive est composée à 99% de bactéries anaérobies
(Bacteroides, Eubacterium, Clostridium, Bifidobacterium, Peptostreptococcus...), la flore sous-
dominante étant représentée par des espèces aéro-anaérobies ou aéro-tolérantes, essentiellement
des entérobactéries type Escherichia coli. Les bactéries anaérobies constituent une véritable flore
de « barrière » qui empêche la prolifération des autres germes. Elles présentent peu de capacités
de translocation 22. Il a été ainsi démontré dans des modèles animaux qu’une destruction sélective
de la flore anaérobie provoquait une prolifération bactérienne dans le tube digestif et facilitait la
translocation des bactéries aérobies 23. Cette prolifération digestive est un élément primordial
dans la translocation bactérienne : il existe par exemple chez la souris un lien entre le nombre de
bactéries présentes dans le tube digestif et dans les ganglions lymphatiques mésentériques 24.
Chez le rat cirrhotique, le nombre de bactéries dans le tube digestif est significativement
supérieur chez les animaux ayant une translocation bactérienne (définie par la présence de
bactéries dans les ganglions lymphatiques mésentériques), qu’elle soit associée ou non à une
infection du liquide d’ascite 25. Chez l’homme, Chang et al ont rapporté une flore microbienne
digestive plus abondante chez les patients cirrhotiques ayant souffert d’ILA par rapport à ceux
n’ayant pas connu d’épisode d’ILA 26. Cette pullulation microbienne pourrait être expliquée par
une diminution de la motilité intestinale. Une étude prospective n’a pourtant pas rapporté de lien
entre pullulation microbienne et survenue d’ILA 27.
Parallèlement, de multiples mécanismes s’opposent à la translocation. En premier lieu, la
muqueuse digestive sécrète une couche glycoprotéique composée de mucines chargées
négativement et qui empêche tout contact étroit entre bactéries et muqueuse. Ces sécrétions sont
également riches en immunoglobulines de type A (IgA) qui participent au phénomène
« d’exclusion » et neutralisent les toxines bactériennes. La bile joue également un rôle par son
action anti-adhérente et anti-toxinique. L’épithélium intestinal est en lui-même une barrière
efficace : les jonctions serrées (tight junctions) au pôle apical des entérocytes permettent le
passage paracellulaire de petites molécules (2 kD) mais empêchent normalement le passage de
bactéries et même de lipopolysaccharides (LPS) bactériens. De plus, il existe une immunité non
spécifique locale, composée de différentes molécules anti-bactériennes comme le lysozyme, les
ou defensines.
Chez le patient cirrhotique, il a été montré diverses altérations de la muqueuse digestive.

- 5 -
Il existe une baisse des défenses immunitaires locales, et en particulier une baisse de la sécrétion
d’IgA, facilitant la pullulation bactérienne. Parallèlement, l’épithélium lui-même est altéré, avec
une augmentation de l’espace intercellulaire. Plus concrètement, le patient cirrhotique a une
perméabilité intestinale augmentée, et ce, d’autant plus que la cirrhose est
avancée 28-31. Campillo et al ont montré que les patients qui développaient une complication
infectieuse (bactériémie et/ou ILA) étaient ceux qui présentaient au préalable une perméabilité
intestinale augmentée 28. Inversement, Ersoz et al n’ont pas trouvé de différence de perméabilité
entre les patients qui souffraient ou non d’ILA au moment de l’épreuve fonctionnelle 29.
La translocation bactérienne au cours de la cirrhose a été étudiée dans différents modèles
animaux 24, 25. Ainsi Llovet et al ont montré que la translocation avait lieu uniquement chez les
animaux cirrhotiques, dans 45% des cas, et que cette translocation était associée dans 60% des
cas à une ILA 32. Dans une autre étude, les germes retrouvés dans l’ascite étaient identiques à
ceux isolés des ganglions mésentériques selon leur profil de restriction après électrophorèse en
champ pulsé 33
.
Garcia-Tsao et al ont montré que la translocation était beaucoup plus fréquente
chez l’animal ascitique que chez l’animal cirrhotique sans ascite 34. Chez l’homme, les études se
heurtent à la difficulté de démontrer la translocation bactérienne en raison de l’impossibilité de
prélever des ganglions mésentériques sans chirurgie. Cependant, dans une large étude prospective
regroupant 101 patients cirrhotiques et 35 témoins non cirrhotiques, tous devant subir une
laparotomie, Cirera et al ont montré que la translocation bactérienne, définie par la présence de
bactéries dans les ganglions lymphatiques mésentériques, était d’autant plus fréquente que la
cirrhose était avancée : le score de Child-Pugh (score de sévérité de la cirrhose) était le seul
facteur de risque indépendant de translocation bactérienne chez le cirrhotique. Ainsi, la
translocation bactérienne a pu être mise en évidence chez 2,6% des témoins, 3,4% des patients au
stade Child A, 8,1% des patients au stade Child B, et 31% des patients au stade Child C. Chez les
patients cirrhotiques au stade Child C recevant une décontamination digestive sélective, la
translocation bactérienne n’a pu être mise en évidence que chez 4,5% d’entre eux 35.
Après translocation, les bactéries sont drainées par le canal thoracique jusqu’à la
circulation sanguine, réalisant une véritable bactériémie qui leur permet d’atteindre l’ascite.
L’ascite a un faible taux de complément, et par conséquent une faible capacité d’opsonisation et
de bactéricidie 36, 37. Un taux faible de complément dans le liquide d’ascite représente d’ailleurs
un facteur de risque d’ILA. L’immunité locale joue un rôle important dans la pathogénie :
l’absence de réaction immunitaire et inflammatoire donne lieu à une simple colonisation souvent
transitoire de l’ascite, appelée bactérascite. A l’inverse, les réactions immunitaires et
inflammatoires sont à l’origine de toute la symptomatologie de l’ILA et des conséquences non
infectieuses, notamment sur la fonction rénale.
 6
7
8
9
10
11
12
13
14
15
6
7
8
9
10
11
12
13
14
15
1
/
15
100%