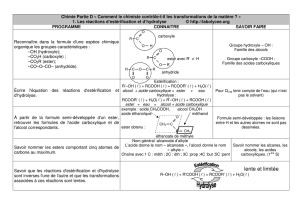
Chimie organique - Option PC - Chapitre 4/5 Acides carboxyliques

Chimie organique - Option PC - Chapitre 4/5
Acides carboxyliques et dérivés d’acide
I Les acides carboxyliques - ions carboxylates :
1. Présentation :
• Un acide carboxylique possède la fonction
carboxyle
Un ion carboxylate possède la fonction carboxylate
• On les trouve à l’état naturel, et ils peuvent être synthétisés au laboratoire
• Caractéristiques physiques :
Teb et Tfus sont assez élevées, car il existe des liaisons hydrogène en nombre
important (plus que dans les alcools de même masse molaire)
acide carboxylique M (g.mol-1)Teb (°C) Teb (°C)M (g.mol-1)alcool
HCO2H
CH3CO2H
CH3CH2CO2H
CH3CH2OH
CH3(CH2)2OH
CH3(CH2)3OH
46
60
74
46
60
74
100,5
118
141
78,3
117,7
97,2
R C O
O H
RC
O
OH
RC
O
OH
R C O
O H
RC
O
OH
et
ils sont solubles dans l’eau, par formation de liaisons hydrogène. Si R- devient de
taille élevée, l’acide n’est plus soluble, car on détruit plus de liaisons hydrogène
H2O/H2O que l’on ne recrée de liaisons hydrogène H2O/R-CO2H
2. Propriétés spectroscopiques :
CO
OCO
OH
fonction
carboxyle fonction
carboxylate

• IR : C=O ≈ 1710 – 1750 cm-1
O-H ≈ 2500 – 3000 cm-1 (signal large, avec < à celui des alcools)
• RMN (H) :
CO
OH
- 10 à 13 ppm
(effet attracteur + effet d'anisotropie)
- 2 à 3 ppm
(effet attracteur du carboxyle)
C
H2CO
O H
variable selon la concentration et le solvant
(liaisons hydrogènes de force variable)
3. Structure et réactivité :
a. Structures des groupes carboxyle et carboxylate :
. Géométries :
alcools, éther-oxydes
acides carboxyliques
122
121
143
134
C=O C-O
R C
O
O H
R C
O
O
d (C/O) (pm)
aldéhydes, cétones
124°
125°
111°
120°
120°
120°
121 pm
134 pm 97 pm
127 pm
acide carboxylique ion carboxylate
. Interprétation : elle est basée sur l’examen des formes mésomères
Pour R-CO2H :

CO
O H CO
O H
majoritaire minoritaire
• d(C=O) ≈ d(C=O)aldéhyde/cétone [121 ≈ 122], car la seconde forme mésomère est minoritaire.
• d(C-O) < d(C-O)alcool/éther [134 < 143], car la seconde forme existe cependant.
Pour R-CO2- :
CO
O
CO
O
formes équiprobables
• d(C/O) est égale pour les deux oxygènes, car les deux formes sont équiprobables.
• d(C=O)aldéhyde/cétone < d(C/O) < d(C-O)alcool/éther [122 < 127 < 143] car la liaison C/O est
alternativement double et simple.
• d(C/O) < d(C-O)acide carboxylique [127 < 134] : le caractère de double liaison est plus fort
dans le carboxylate que dans l’acide carboxylique, car la seconde forme de l’acide est
minoritaire.
b. Réactivité :
• Acidité de H dans -CO2H :
P(O) > P(H) : l’hydrogène est donc H+, et a un caractère acide.
La base carboxylate est stabilisée par mésomérie, plus que ne l’est le groupe -CO2H.
La base carboxylate est donc particulièrement stable.
-CO2H perd donc facilement un proton. C’est un acide de Brønsted
• Basicité de -CO2H :
-CO2H peut capter un proton par l’intermédiaire d’un doublet libre, sur
l’ou ou l’autre des oxygènes. Il existe deux sites de protonation possibles,
numérotés (1) et (2) :
CO
O H
(1)
(2)

CO
O H
HCO
O H
H
CO
O H
H
Site (1)
Site (2)CO
O H
H
On aura donc une protonation préférentielle sur le site (1).
• Acidité du H sur le C en
du carbonyle :
C C O
O H
H
moins acide que le H en d'un carbonyle
Le groupe -OH a un effet +M, qui diminue l’effet attrateur du carbonyle du carboxyle
C’est le H acide du groupe carboxyle qui réagira préférentiellement avec une base.
• Ccentral peu électrophile :
Le groupe -OH a un effet +M, qui diminue l’effet attrateur du carbonyle du carboxyle. Donc,
le carbone du carbonyle est moins électrophile.
Le carboxyle devra donc être activé pour pouvoir subir l’attaque de nucléophiles.
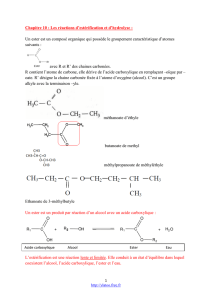
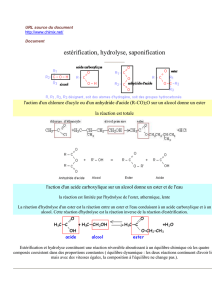
II Synthèse des esters et autres fonctions dérivées :
1. Présentation des fonctions dérivées :
a.R C O
Z
:

Cl
O C O
R'
O R'
NR1
R2
chlorure d'acyle
anhydride d'acide
ester
amide
Me C O
Cl
Me C O
O
C
O
Et
Me C O
O CH3
Me C O
NCH3
CH3
chlorure d'éthanoyle
anhydride d'acide éthanoïque
et propanoïque
éthanoate de méthyle
N,N-diméthyléthanamide
Z
chlorure d’acyle et anhydride d’acide : produits de synthèse exclusivement
ester et amide : synthétisés au labo, ou trouvés à l’état naturel
b.R C :
N
Ce sont des nitriles, qui sont des produits de synthèse essentiellement
H3C CH2C N propanenitrile
2. Structure et réactivité de R C O
Z:
a. Structure :
Z est conjugué avec le carbonyle de la fonction carboxyle :
R C O
ZR C O
Z
Le doublet porté par Z diminue donc globalement le pouvoir électrophile du C central sp2.
Cependant, ce carbone central peut subir l’attaque de nucléophiles.
b. Influence de la nature de Z :
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%