Sarcomes Tissus Mous

OPIO SFCP MARS 2009
RESUME FAIT PAR ROGER MOUAWAD (Paris-AERIO)
SARCOME DES TISSUS MOUS
ROLE DE LA CHIMIOTHERAPIE ADJUVANTE DANS LES SARCOMES DES TISSUS MOUS EN 2009
Docteur Axel LECESNE, Institut Gustave Roussy, Villejuif
L’incidence estimée des sarcomes des tissus mous est de 19 cas par million d’habitants et par an. La
mortalité est de 50 % environ, essentiellement par le fait d’une dissémination métastatique. Le risque
de rechute locorégionale est de 20-30 %. Le pronostic est aggravé en cas de récidive. La prise en
charge des sarcomes des tissus mous (STM) est en pleine évolution du fait des progrès effectués
dans les classifications biologiques, cytogénétiques et dans l'essor des thérapeutiques ciblées qui
modifient peu à peu l'arsenal et les stratégies thérapeutiques dans ce domaine.
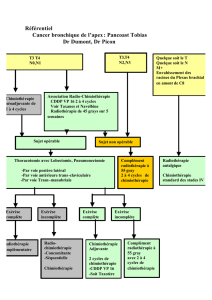
Traitements
Une chirurgie optimale initiale demeure toujours l'acte thérapeutique principal et incontournable des
sarcomes des tissus mous de haut grade localisé opérable. Compte-tenu de l'efficacité croissante des
thérapeutiques non chirurgicales, celle-ci doit cependant systématiquement s'intégrer dans le cadre
d'une approche pluridisciplinaire intégrant dans la stratégie thérapeutique la localisation tumorale, son
profil évolutif, le grade histologique, la taille tumorale, le sous-type histologique et l'âge du patient.
Dans ce contexte, la place de la chimiothérapie adjuvante a souvent été discutée. Dans une méta-
analyse portant sur 13 études randomisées (plus de 1500 patients) posant la question de l'intérêt de la
chimiothérapie adjuvante à base d'anthracyclines associée à un alkylant démontrait un bénéfice
significatif de 10 % à 5 ans sur la survie sans récidive mais pas sur la survie globale. Les analyses par
sous-groupes effectuées montrent que les hommes de 30 à 60 ans et les sarcomes des tissus mous
(sauf les léiomyosarcomes) des extrémités de 5 à 10 cm bénéficient le plus de la chimiothérapie.
Pour les sarcomes des tissus mous des extrémités de haut grade, une étude randomisée menée
sur 104 patients démontraient un bénéfice de la chimiothérapie adjuvante (Ifosmamide/ epirubicine)
sur la survie sans récidive et la survie globale. Malheureusement, l'impact positif de la chimiothérapie
sur la survenue de métastases s'estompe dans le temps, soulignant l'importance d'un suivi prolongé
des patients opérés d'un sarcomes des tissus mous de haut grade de malignité. Enfin, l’étude de
l'EORTC, incluant 351 patients sur 9 ans, et présentant un STM de haut grade de toute localisation (y
compris les sarcomes viscéraux), n'a montré aucun bénéfice de la chimiothérapie
(Doxorubicine/lfosfamide) aussi bien sur la survie sans récidive que sur la survie globale.
Au regard des enjeux thérapeutiques et de la complexité de cette pathologie, il a paru indispensable
d'aller plus loin dans l'objectif d'identifier des sous-groupes susceptibles de bénéficier le plus des
apports de la chimiothérapie adjuvante. Une analyse détaillée des 2 études consécutives de l'EORTC
présentée à l'ASCO 2008 montre que, la qualité de la résection initiale, le grade histo-pronostique et
la taille tumorale représentent des facteurs pronostices indépendants pour la survie, quel que soit le

traitement administré. Par contre, le sexe, la qualité de la résection (exérèse R0 versus R1 ) et l'âge (±
40 ans) sont des facteurs prédictifs indépendants de chimio-sensibilité pour la survie globale. Enfin,
les hommes, les patients de plus de 40 ans et les patients qui ont été opérés initialement de façon non
optimale (exérèse R1) bénéficient le plus d'une chimiothérapie adjuvante (anthracyclines / ifosfamide).
La survie globale à 10 ans des patients ayant bénéficié d'une exérèse incomplète passe ainsi de 27.6
% sans chimiothérapie à 44.6 % avec une chimiothérapie adjuvante. Par contre cette dernière ne
modifie pas la survie globale à 10 ans des patients opérés de façon optimale. (60.6% sans
chimiothérapie vs 57.7 % avec).
La chimiothérapie adjuvante améliore ainsi la survie globale des patients n'ayant pas bénéficié d'une
prise en charge chirurgicale initiale mais ne la remplace aucunement (44.6 % à 10 ans après exérèse
R1 avec chimiothérapie, 60.6 % à 10 ans après exérèse R0 sans chimiothérapie). Le pronostic du
patient ayant un sarcome des tissus mous localisé commence au stade initial de sa prise en charge et
son suivi doit être poursuivi au-delà de 5 ans. Aucun sous-type histologique ne se démarque de cette
analyse. Les patients de plus de 40 ans bénéficient le plus de la chimiothérapie adjuvante pour des
raisons encore obscures, non liées à une plus mauvaise prise en charge initiale (type d'exérèse
identique chez les jeunes et moins jeunes) ni aux caractéristiques des sarcomes des tissus mous
dans ces 2 populations. Enfin, comme dans la méta-analyse, ce sont les hommes qui bénéficient le
plus de la chimiothérapie adjuvante.
En conclusion, l'intérêt d'une chimiothérapie adjuvante doit désormais être discuté au cas par cas
dans des comités pluridisciplinaires. Si elle semble requise en cas d'exérèse R1, elle est plus
discutable après un traitement locorégional optimal. Elle s'impose certainement plus en situation
néoadjuvante (après exérèse R1 et R2) avant une reprise chirurgicale large qu'en situation adjuvante
à tous les patients. Si elle est utilisée, la chimiothérapie doit comporter une anthracycline
(Doxorubicine, 60 à 75 mg/m² au Jl) et l'ifosfamide administrée selon un schéma fractionné sur
plusieurs jours pour une dose totale moyenne de 9 g/m².
FACTEURS PREDICTIFS DE REPONSE
Pr Jean-Yves BLAY
Introduction
Le sarcome des tissus mous (STS) est une tumeur rare (représentant moins de 1 % des tumeurs de
l’adulte). On estime l’incidence annuelle en France à 2 500 nouveaux cas. Tous les âges sont
concernés, sans prédominance géographique ni ethnique. Le sexe ratio est équilibré. Il s’agit d’un
groupe tumoral hétérogène comportant plus de 50 sous-types histologiques, associés parfois à des
pronostics différents. Après la prise en charge d’un sarcome localisé, les patients sont exposés au
risque de récidive locale et de dissémination secondaire pulmonaire. Dans la plupart des séries, le
taux de survie des STS au stade avancé est de 12 mois au mieux et le taux de réponse objective à la
chimiothérapie reste faible, de l’ordre de 25-40 %. L’identification des facteurs pronostics et prédictifs
de réponse aux thérapeutiques constitue un véritable défi, et ce, afin de mieux comprendre la biologie
de ces tumeurs et d’optimiser les thérapeutiques. Pour les sarcomes, les facteurs prédictifs de

réponse bien établis sont essentiellement liés à la tumeur : associent le type histologique, le site des
métastases, la taille de la tumeur, le sous type moléculaire, mutations primaires et secondaires ; liés
au patient : l’âge au diagnostic, le sexe et le génotype. Outre ces facteurs cliniques classiques, la mise
en évidence d’une anomalie moléculaire activatrice ou récurrente dans un sous-type histologique
donné s’est montrée bien plus efficace pour l’élaboration d’un traitement que la simple expression
d’un marqueur. Grâce à l’évolution des connaissances en matière de biologie des sarcomes, on a pu
identifier des facteurs pronostics et prédictifs moléculaires qui ont permis le développement de
traitements spécifiques adaptés à chacun des sous-type histologique.
Marqueurs moléculaires
De très nombreuses altérations génomiques ont été observées et les sarcomes sont actuellement
subdivisés en entités moléculaires et histologique bien distinctes. Six sous types de tumeurs
conjonctives ont ainsi été identifiés, en fonction des anomalies moléculaires rencontrées : 1)
translocations spécifiques aboutissant à la constitution de gènes de fusion codant des modulateurs de
transcription ou des protéines qui agissent comme des facteurs de croissance (Ewing t(11;22) ,
Synovialosarcomas t (X;18), Alveolar rhabdomyosarcomas t(1;13), t(2;13) ; DSRCT t(11;22) etc.. 2)
mutations touchant les récepteurs à tyrosine kinases (KIT et PDGFRA dans les GIST); 3) délétion de
gènes suppresseurs de tumeur (NF1 impliqué dans la neurofibromatose de type 1 ou INI1 dans les
tumeurs rhabdoïdes); 4) altérations génétiques simples (amplification de mdm2/cdk4 dans les
liposarcomes bien différenciés ou dédifférenciés) 5) altérations génétiques plus grossières (comme
dans le cas du leiomyosarcome); 6) altération des voies de l’adhésion cellulaire (délétion du gène
-catenine dans les fibromatoses agressives).
Nouvelles stratégies thérapeutiques
Elles suivent le même principe de ciblage spécifique des différents sous-types moléculaires et
histologiques. Le ciblage de KIT par des inhibiteurs spécifiques de tyrosine kinase (imatinib, sunitinib,
et plus récemment nilotinib) est le premier traitement ciblé ayant démontré un gain de survie chez des
patients résistants à la chimiothérapie et à la radiothérapie. L’étude (ACOSOG-Z9001) compare
l’imatinib à la dose de 400 mg/jour pendant un an versus placebo, chez des patients recevant un
traitement adjuvant par imatinib après l’exérèse (R0/R1) d’un GIST de plus de 3 cm. La survie sans
récidive à un an était de 97% dans le bras imatinib contre 84% dans le bras placébo (p < 0.001) avec
un bénéfice de l’imatinib s’observant principalement dans les GIST opérés ayant une taille de plus de
6cm (p=0.01) et de plus de 10 cm (p<0.001) où la survie sans récidive à un an est de 55% dans le
bras placébo contre 95% dans le bras imatinib. La différence n’est pas significative pour les GIST de
petites tailles (de 3 à 6 cm). Dans ces stades avancés, les recommandations actuelles préconisent
l’utilisation d’imatinib mésylate (400 mg/jour) en première ligne de traitement. Chez les patients en
progression, l’imatinib à la dose de 800 mg/jour permet le contrôle de la tumeur dans 30 à 35 % des
cas, avec une survie sans progression de 4 mois en moyenne. Les résultats d’une méta-analyse de
deux études randomisées comparant les deux niveaux de dose d’imatinib (400 mg/jour vs. 800
mg/jour) démontre a supériorité de la plus forte dose, avec un allongement de la survie sans
progression chez les patients porteurs de mutations de l’exon 9. Ce qui fait de la mutation dans les

Gist le facteur pronostique le plus important pour la sensibilité ou la résistance à l'imatinib. A l’issue
de l’ESMO 2008, l’utilisation de l’imatinib à 800 mg/jour et considéré comme le traitement standard de
première ligne dans cette indication. L’étude BRF14 a cherché à évaluer si l’interruption du traitement
était envisageable après 1 ou 3 ans de traitement. Les résultats ont montré un taux >90 % de
progression chez les patients atteints de GIST avancés avec une médiane de survie sans progression
de 6 mois après l’interruption du traitement. Même si la reprise de l’imatinib a permis d’obtenir un
nouveau contrôle de la maladie chez la plupart des patients, l’impact de cette interruption sur la survie
globale reste à déterminer.
La voie RANK/RANK ligand et incontestablement le scoop dans les tumeurs à cellules géantes
osseuses localement avancées ou récidivantes. Ces tumeurs stimulent les ostéoclastes via la voie
RANK/RANK ligand. Le denosumab se fixe au récepteur RANK avec une grande affinité. Administrée
à la dose 120 mg en sous-cutané tous les 28 jours, les résultats se passent de commentaires,13
réponses sur 15 patients actuellement évaluables (87%) soit en terme de réponse histologique (9/9) et
(4/6) en terme de réponse radiologique, diminution de la destruction osseuse, reformations d’une
structure osseuse normale, réponse métabolique au PET. Pour être définitivement complet un rôle
possiblement non négligeable de la voie IGFR1 qui est non seulement activée dans les GIST wild type
mais également dans les PNET et possiblement dans les sarcomes des tissus mous à génétique
simple sous-tendus par des translocations chromosomiques spécifiques. Les études sur l’inactivation
de la voie IGFR1 à l’aide d’anticorps monoclonaux sont en cours. L’étude SARC011 est certainement
la plus avancée avec plus de 100 patients inclus et des réponses spectaculaires dans des sarcomes
d’Ewing métastatiques lourdement prétraités ont été montrées. En ce qui concerne les cytotoxiques,
certains agents ont une efficacité spécifique reconnue, comme la trabectedin-ET743 (Yondelis®) qui a
obtenu l’AMM dans les STM métastatiques après échec d’une chimiothérapie à bases
d’anthracyclines et d’ifosfamide. Enfin, l’analyse du profil d’expression génique et l’analyse par
microarrays ont montré que plusieurs sous-types histologiques de sarcomes, en particulier les
sarcomes synoviaux et les tumeurs des gaines nerveuses périphériques, expriment fortement HER1.
Des études de phase II avec des inhibiteurs de tyrosine kinase non pas montrés d’efficacité dans ces
indications.
Conclusion
Grâce aux progrès considérables accomplis en matière de caractérisation biologique de ces tumeurs,
des nouvelles stratégies thérapeutiques utilisant des agents cytotoxiques ciblant spécifiquement
certains sous-types histologiques ont pu se développer. Ces traitements ont permis des avancées
importantes tant pour le contrôle de la maladie que pour la survie des patients. L’évolution constante
des techniques de diagnostic moléculaire devrait permettre d’adapter le traitement des GIST à leur
statut mutationnel. Actuellement, le traitement prend en compte les mutations éventuelles de KIT et
PDGFR. Dans les autres sous-types de sarcomes des tissus mous, la généralisation des techniques
de caractérisation moléculaire et la stratification des patients selon leur sous-type histologique telle
qu’elle est pratiquée dans les essais cliniques récents devraient déboucher sur une partition des GIST

en plusieurs entités distinctes bénéficiant de traitements différents.
RADIOTHERAPIE DES SARCOMES DES TISSUS MOUS
Docteur Yazid BELKACEMI, Hôpital Henri Mondor, Créteil.
Les sarcomes des tissus mous font partie des pathologies malignes rares, moins de 1 % de la totalité
des cancers. Ils doivent bénéficier d'une prise en charge spécifique par des centres de référence. Leur
incidence avoisine 1500 nouveaux cas par an siégeant dans 2/3 des cas au niveau des membres.
Place de la radiothérapie dans la stratégie thérapeutique
L'association radio-chirurgicale conservatrice reste le traitement standard des formes opérables
localisées aux extrémités. De nombreux essais ont permis de montrer l'impact positif de la
radiothérapie. Cependant, quelques avantages et inconvénients peuvent être notés selon que cette
radiothérapie a été réalisée en pré ou post-opératoire.
La radiothérapie pré-opératoire à la dose de 50 Gy a comme inconvénients de ne pas disposer de
matériel diagnostic important, de ne pas pouvoir évaluer la réponse à une éventuelle chimiothérapie et
d'augmenter le risque de complications aiguës et le retard à la cicatrisation. La radiothérapie post-
opératoire est, quant à elle, administrée de façon plus retardée à des doses plus élevées et dans des
volumes plus importants avec un risque plus élevé de complications tardives et de séquelles
fonctionnelles.
Apport de la radiothérapie de haute technicité
Actuellement, la radiothérapie des sarcomes des membres est réalisée selon les techniques
conformationnelles tri dimensionnelle (RTC 3D). Nous ne disposons pas encore de données
suffisantes pour la généralisation de la radiothérapie avec modulation d'intensité (RCMI), une
technique indéniablement prometteuse pour le traitement des sarcomes des extrémités. En revanche,
pour de multiples autres localisations (tête et cou, prostate et autres localisations profondes), la RCMI
permet d'augmenter l'homogénéité de la dose dans le volume cible tout en épargnant mieux les
organes sains avoisinants. La RCMI peut être réalisée par un accélérateur linéaire traditionnel ou
grâce aux machines de haute technicité comme la Tomothérapie. Cette dernière associe un
accélérateur linéaire à un scanographe hélicoïdal. Elle permet de combiner les avantages de la RCMI
et à ceux de la radiothérapie guidée par l'imagerie (IGRT) pour des bénéfices dosimétriques nets par
rapport à la RTC 3D. Lors de l'irradiation des sarcomes des extrémités par exemple, cette modalité
permet une meilleure maîtrise des difficultés rencontrées avec les larges volumes situés à proximité
du membre controlatéral et des organes génitaux externes.
En conclusion, pour le traitement des sarcomes des extrémités, la technique conformationnelle tri
dimensionnelle est utilisée en routine. Le respect du membre controlatéral limite fortement le nombre
de faisceaux utiliser pour couvrir correctement le volume cible sans exposer les tissus sains
environnants. L'enjeu de la radiothérapie de haute technicité sera de maintenir voir d'augmenter le
 6
6
1
/
6
100%