File - Cours L3 Bichat 2012-2013

1/10
UE5 : génétique médicale
Le 5/11/12 à 8h30
Pr E.Tournier-Lasserve (responsable de l’UE)
Ronéotypeuse : Aurore Protat
Ronéolecteur : Karim Abdelmoumen
Cours 1 : Hérédité Mendélienne

2/10
SOMMAIRE :
RAPPELS
I/ MALADIES AUTOSOMIQUES DOMINANTES
II/ MALADIES AUTOSOMIQUES RECESSIVES
III/ MALADIES RECESSIVES LIEES A L’X
IV/ MALADIES MITOCHONDRIALES
V/ L’EMPREINTE GENOMIQUE
Réf : cours de génétique médicale du collège des enseignants de génétique (Masson)/ biologie
moléculaire et médecine (Flammarion 3ème édition 2007)
Précisions pour les ED : Les 3 ED porteront sur des exemples de maladies génétiques qui n’auront pas
été abordées dans le cours ou juste effleurées. Il faut RESPECTER LES GROUPES car il y aura une liste
d’émargement au début de l’ED. A la fin de l’ED, une question de 15min dans le cadre du contrôle
continu qui représentera 20% de la note finale. ATTENTION ! pour que votre copie soit prise en
compte il faut que vous soyez dans la bonne salle et à la bonne heure.

3/10
RAPPELS
Définition :
Un gène= séquence nucléotidique qui code pour une ou plusieures protéines/ unité d’information
génétique
Un locus= emplacement du gène sur le chromosome
Un allèle= version alternative d’un même gène qui diffère par sa séquence nucléotidique, on possède
2 allèles par gène. Ces variations pour un gène donné sont responsables de la diversité des
phénotypes et ne sont pas nécéssairement pathologiques.
Hétérozygote= individu qui porte 2 versions différentes (allèle) d’un même gène sur chacun de ses
chromosomes homologues
Hémizygote= individu qui ne posséde qu’une seule copie d’un même géne ; exemple tout à fait
physiologique : les individus XY ne présentent qu’une seule copie des gènes présents sur les
chromosomes sexuels. Parfois l’hémizygotie aboutit à des pathologies.
Hétérozygote composite= individu qui présente 2 alléles mutés d’un même gène mais dont les
mutations sont différentes
Homozygote= individu qui possède le même allèle sur chacun de ses 2 chromosomes homologues
Allèle dominant (A) et allèle récessif (a) :
Un individu est porteur du génotype A//a: - s’il présente le phénotype [A] alors on peut dire que
l’allèle A est dominant. Pour que l’individu présente le phénotype [a] il faut donc qu’il possède le
génotype a//a.
On parle de semi-dominance lorsqu’un individu A//a présente un phénotype intermédiaire entre les
phénotypes [A] et [a].
On parle de co-dominance si les 2 allèles A et a s’expriment en même temps, c'est-à-dire qu’on
retrouvera à la fois la protéïne A et la protéïne a (ex : groupes sanguins ABO).
Nature des mutations :
→structure d’un gène (ADN génomique) : un gène est organisé en une suite d’introns et d’exons.
-un exon est une séquence nucléotidique que l’on retrouve dans l’ARNm, attention il n’est pas
nécéssairement traduit en protéïne ; il y a des exons condants, non codants ou partiellement codants
-un intron est une séquence absente de l’ARNm. Ils se situent entre les exons. Ils contiennent les
sites consensus d’épissage (dans les régions directement en amont et en aval des exons). En amont
du premier exon on retrouve une zone de régulation.
-l’épissage alternatif est la transcription d’exons différents qui permet d’obtenir différents ARNm
(donc différentes protéïnes) pour un même gène.
Si des mutations se produisent dans ces régions cela aboutit souvent à des pathologies.
→ mutation non sens : mutation ponctuelle dans la séquence nucléotidique qui change un codon qui
code pour un acide aminé en un codon STOP. (mutation dans un exon)
Mutation faux sens : mutation ponctuelle qui change un codon qui code pour un acide aminé en un
en un codon qui code pour un autre acide aminé. (Mutation dans un exon)
Mutation dans un site d’épissage : la machinerie de transcription ne reconnait plus le site consensus
et va « sauter » au prochain site d’épissage : on perd un exon dans l’ARNm. Si l’exon manquant est

4/10
un multiple de 3, on n’aura pas de décalage du cadre de lecture ; la protéine sera tronquée, si ce
n’est pas un multiple de 3 le cadre de lecture sera décalé ; apparition prématurée d’un codon STOP.
Insertion/délétion de nucléotides : - cela se produit dans un exon et le gain/perte de nucléotides est
multiple de 3 ; il n’y a pas de décalage du cadre de lecture, il y a quelques acides aminés en plus/en
moins dans la protéine ;
- cela se produit dans un exon et le gain/perte de nucléotides n’est
pas un multiple de 3 ; il y a un décalage du cadre de lecture, dans la majorité des cas cela aboutit à
l’appartion d’un codon STOP prématuré.
→Les conséquences de ces mutations : - les mutations faux sens n’ont pas toujours de conséquences
fonctionnelles : selon la position de l’acide aminé muté dans la protéine.
- les mutations non sens/ les décalages du cadre de lecture/
les mutations dans les sites d’épissage ont dans ≈100% des cas des conséquences fonctionnelles : la
protéine est anormale, tronquée, non fonctionnelle voir absente.
Toutes les spécialités sont concernées par la génétique, certaines plus que d’autres (pédiatrie,
neurologie). Il faut acquérir quelques bases notament il faut savoir dessiner un arbre généalogique et
déterminer l’origine génétique d’une maladie :
-les hommes sont des carrés/ les femmes sont des ronds
-on représente les parents du premier degrès (parents, frères et sœurs, enfants) si c’est possible,
ceux du 2ème degrès (oncle et tante, grands parents)
-il faut dessiner les fausses couches et les morts-nés
-on note l’AGE ET LA CAUSE DE DECES
-on note toutes les maladies présentes dans la famille
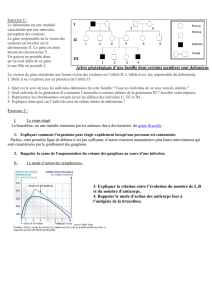
Pour déterminer le mode de transmission de la maladie et ainsi calculer le risque de récurrence, il
faut regarder le sexe de l’enfant atteint et le sexe du parent transmetteur ainsi que le pourcentage
de sujets atteints dans l’arbre
Dans ce cours on s’intéressera aux maladies mendéliennes (=monogénique) mais il existe aussi des
maladies polygéniques multifactorielles très fréquentes.
I/ MALADIES AUTOSOMIQUES DOMINANTES
50% des maladies mendéliennes
Un individu atteint d’une maladie autosomique dominante possède un allèle muté (m) et un allèle
normal (+) : il est hétérozygote. Lors de la gamétogénèse les chromosomes homologues se séparent
et l’individu va produire 50% de gamètes qui portent l’allèle (m) et 50% qui portent l’allèle (+).
Comme il s’agit de maladies rares le plus souvent le conjoint est normal. L’enfant aura 50% de risque
de recevoir l’allèle (m) et donc d’être malade et 50% de chance de recevoir l’allèle (+).

5/10
Caractéristiques d’une maladie autosomique dominante :
- en général le sujet atteint possède un parent atteint
- le sujet atteint transmet statistiquement sa maladie à 50% de sa descendance
- peut être transmise aussi bien par le père que par la mère
- les enfants atteints sont aussi bien des garçons que des filles
Cependant la réalité clinique est plus complexe :
-Si un malade fait des enfants avec un aute malade : c’est une situation rare (sauf dans le cas des
déficits sensoriels : surdité etc, car les patients fréquentent des associations de malades où ils
rencontrent leur conjoint). Dans ce cas le phénotype M//M est parfois le même que M//m ou parfois
plus grave.
-Mutation de novo : aucun parent n’est atteint. La mutation peut se produire dans les gamètes de
l’un des 2 parents ou très tôt au cours de l’embryogénèse ; dans certaines maladies les mutations de
novo représente 100% des malades car ils n’ont pas de descendance, c’est le cas du nanisme
achondroplasique. Evidement le sujet pourra transmettre cette mutation à sa descendance.
-Maladie à pénétrance incomplète : un individu est porteur de l’allèle muté (m) mais ne
développe pas la maladie, elle se définit comme le nombre de sujets qui expriment la maladie/le
nombre de sujet porteurs de la mutation. La pénétrance est âge dépendante à moins que les
symptomes apparaissent lors des premiers jours de vie. Très souvent elle est incomplète. Par
exemple le carvernome cérébral a une pénétrance de 60% et la CADASIL (cerebral autosomal
dominant arteriopathy with subcortical infarcts and leukoencephalopathy) de 100%. Le caractère
incomplet de la pénétrance est responsable des « sauts de générations » car même s’il n’a pas de
symptômes de la maladie le sujet peut la transmettre à sa descendance
-L’expressivité est variable : elle représente la variabilité des phénotypes des patients. Prenons
l’exemple de la maladie de Marfan (revue pendant les Ed) ; c’est une maladie rare (≤1/10000) il s’agit
d’une mutation dans le gène de la fibrilline qui est responsable d’anomalies du tissu conjonctif. Le
phénotype est variable avec des symptômes qui vont de l’hyperlaxité ligamentaire à l’anévrisme de
l’aorte. La neurofibromatose (revue en Ed) fait partie des maladies autosomiques dominantes les plus
fréquentes, elle présente une large expressivité : de la tâche café au lait à l’énorme fibrome.
-Problème de la mosaïque gonadique : les 2 parents sont sains (maladie à pénétrance complète)
mais ils ont plusieurs enfants atteints ; face à cela on pose la question qui fâche : est ce qu’il s’agit
bien des enfants biologiques du couple? Si oui, on recherche une mutation dans le génome des
globules blancs mais on ne trouve rien. On va chercher dans les gonades et on trouve des cellules
germinales mutées. Il s’agit d’une mutation de novo qui a eu lieu dans les cellules germinales du
parent ; le % de cellules mutées dépend du stade de la gamétogénèse où s’est produite la mutation.
Contrairement aux mutations de novo il y a un risque de récurrence pour les enfants suivants.
-Le phénomène d’anticipation : il concerne les maladies à triplets ; la mutation est instable, le
nombre de triplets responsables de la maladie augmente à chaque génération ; donc à chaque
génération la maladie est plus précoce ; parfois les parents meurent avant le début de leur maladie
(voilà pourquoi il faut connaître l’âge de décès)
En génétique on se retrouve souvent face à plusieures hypothèses, il faut faire attention à ne pas
tout confondre avec une mutation de novo.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%