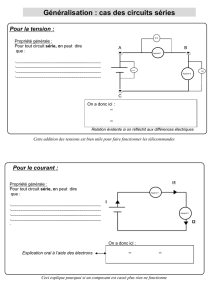
these the final

UNIVERSITE DE NANTES
FACULTE DE MEDECINE
ETUDE
DES
FACTEURS
MODULANT
LE
TRANSFERT
DE
GENE
DANS
LE
MUSCLE
SQUELETTIQUE
DE
PRIMATE
A
L’AIDE
D’UN
AAV
RECOMBINANT
THESE DE DOCTORAT
Ecole doctorale : Chimie-Biologie
Discipline : Médecine
Spécialité : Virologie
présentée
et soutenue publiquement par
TOROMANOFF Alice
le 31 Octobre 2008, devant le jury ci-dessous
Président du jury : Dr. Ignacio Anegon
Rapporteurs : Pr. Serge Herson
Pr. Jean-Christophe Pages
Examinateur : Pr. Fatima Bosch
Directeur de thèse : Dr. Philippe Moullier

A mes parents
A mes frères et sœurs
A ma toute ma famille
A Olivier et Oriane.

R
EMERCIEMENTS
Je souhaite tout d’abord remercier Philippe Moullier, directeur de l’unité INSERM 649,
pour m’avoir accueillie dans son laboratoire et au sein de son équipe.
Je tiens également à remercier Caroline Le Guiner, pour m’avoir prise sous son aile dès
mon arrivée au laboratoire en DEA, et pour m’avoir ensuite encadrée tout au long de cette
thèse. Merci Caroline pour ta disponibilité, ton soutien quotidien, tes conseils précieux et ton
amitié.
Je remercie Monsieur Serge Herson et Monsieur Jean-Christophe Pages pour avoir accepté
d’être les rapporteurs de cette thèse.
Je remercie Madame Fatima Bosch d’avoir accepté d’être examinateur de cette thèse.
Je remercie Monsieur Ignacio Anegon, qui m’a initiée à l’immunologie et qui m’a suivie
tout au long de travail de thèse, d’avoir accepté d’être président du jury de cette thèse.
Je remercie également les personnes extérieures au labo avec lesquelles j’ai travaillé
directement ou collaboré : Yan Cherel, Lydie Guigand, Thibaut Larcher, Jack-Yves
Deschamps, Marcello Hill, Olivier Gauthier, Guillaume Podevin, pour leur aide
indispensable, leur disponibilité, leurs conseils et leur intérêt pour le sujet.
Je remercie vivement tout le personnel du centre de Boisbonne, sans qui ce travail n’aurait
pas été possible, et particulièrement David, Anthony, Mickael, Vanessa, Luc, Arnaud,
Margot, Amandine, Laetitia et Corinne. Merci d’avoir pris soin de mes petits macaques et de
mes souris !!

Un grand merci à toute l’équipe du LAV, et particulièrement aux AAVettes pour les
nombreuses productions de vecteurs…
Je remercie vivement tous les membres du laboratoire de Thérapie Génique pour leur
soutien et leur convivialité, en particulier ceux de mon équipe : ma petite Aurélie qui me
manque déjà ainsi que nos conversations (scientifiques… si si !) dos à dos, Sylvain, Carine,
Béatrice, Magalie, Mickaël (pour tout le boulot que je t’ai donné en PCR et que tu as toujours
fait dans la bonne humeur, un merci particulier !), Delphine, Mia, Adrien, et Gwladys la petite
dernière. Merci pour votre bonne humeur, cela a été un plaisir de travailler avec vous tous !
Merci à Anita, Françoise, Manuela, Cynthia et Charlène.
Merci également…
A Céline, ex-U533, pour tous nos midis passés ensemble au self et pour ta bonne humeur.
Vous étiez les derniers irréductibles Nantais…
A tous mes amis extérieurs au laboratoire, et particulièrement à So et Ol. Malgré la
distance nous pouvons toujours compter les uns sur les autres ! J’en profite pour saluer Arthur
mon filleul préféré !
A ma famille. Vous avez toujours su me soutenir et m’encourager. Merci particulier à mes
parents, à mes frères et sœurs Nicolas, Cathia, Anne-Laure, Marion et Alexis, à Yvette, a mes
oncles, tantes et cousins de Troyes.
A ma belle-famille.
A Olivier. Merci de tout cœur de croire en moi et de m’avoir soutenue à ta manière malgré
tes nombreux déplacements… Merci d’être « près de moi » tout simplement… Et bien sûr,
merci de m’avoir donné ORIANE, notre adorable petite fille ! Ma puce, je te dédie cette
thèse ! Sache que ça n’a pas toujours été facile d’être maman tout en écrivant cette la thèse,
mais nous y sommes arrivés…et tu peux être fière de toi car tu as su être calme et patiente
lorsque je n’avais pas beaucoup de temps à te consacrer… Mais dès à présent, on va rattraper
le temps perdu !!

Liste des abréviations
AAT : alpha-1-antitrypsine
AAV : adeno-associated virus
AAVr : recombinant adeno-associated virus
ADN : acide désoxyribonucléique
ADNc : ADN complémentaire
ARNm : acide ribonucléique messager
BMD : Dystrophie musculaire de Becker
CAG : promoteur chimère de la β-actine de poulet fusionné à l’enhancer du promoteur précoce du
Cytomégalovirus
CD : cellule dendritique
cm : macaque cynomolgus (Macaca fascicularis)
cmEpo : érythropoïétine de macaque cynomolgus
CMH: complexe majeur d'histocompatibilité
CMV : Cytomégalovirus
CPA : cellule présentatrice d’antigènes
CsCl : chlorure de césium
CTLA4 : cytotoxic T lymphocyte-associated protein 4
db : double brin
DMD : Dystrophie musculaire de Duchenne
Dox : doxycycline
ELISA : Enzyme-Linked ImmunoSorbent Assay
Epo : érythropoïétine
FIX : facteur IX de la coagulation
GFP : green fluorescent protein
GMP : Good Manufacturing Procedure
gsg : γ-sarcoglycan
HIV : Human Immunodeficiency Virus
HSV : Herpes Simplex virus
Ht : hématocrite
IFN : Interféron
Ig : Immunoglobuline
IL : interleukine
IM : intramusculaire
ITR : inverted terminal repeat
IV : intraveineux
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
1
/
201
100%