Nouvelles cibles thérapeutiques de l`insuffisance

PHYSIOPATHOLOGIE CARDIOVASCULAIRE
34 AMC pratique n°229 juin 2014
© 2014 Elsevier Masson SAS. Tous droits réservés
La publication de nouvelles connais-
sances sur les aspects moléculaires et
cellulaires du remodelage cardiaque
conduisant à l’insuffisance cardiaque, aussi
bien systolique que diastolique, et aux
arythmies qui lui sont associées est inces-
sante et il est bien de faire le point de
temps en temps sur les principales acquisi-
tions et les principales tendances. Le dernier
« Printemps de la cardiologie » qui s’est
déroulé à Strasbourg les 24 et 25 avril en a
d’ailleurs donné l’occasion. Le remodelage
cardiaque concerne aussi bien les myocytes
que les autres types de cellules du myocarde
ainsi que la matrice extracellulaire. Je me
limiterai dans cette courte revue à l’insuffi-
sance cardiaque systolique et aux altéra-
tions des flux calciques du myocyte
cardiaque qui revêtent un aspect véritable-
ment central dans la dégradation de sa
fonction contractile et, au-delà, dans son
remodelage et sa survie. Ceci m’amènera à
passer sous silence bien d’autres cibles
thérapeutiques potentielles comme, par
exemple, d’autres aspects du dysfonction-
nement du myocyte tels que les altérations
de son métabolisme énergétique et de ses
mécanismes de survie ou encore celles
permettant de moduler la régénération
myocardique à partir de divers types de
cellules.
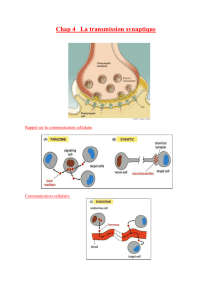
Altérations des flux calciques
au cours de l’insuffisance
cardiaque systolique
Il est maintenant admis que dans l’insuffisance
cardiaque systolique avancée, l’altération du
fonctionnement du réticulum sarcoplasmique
(RS) joue un rôle central. Elle est le fait de la
diminution du recaptage du Ca2+ dans le RS
pendant la diastole et d’une fuite du Ca2+ hors
du RS également pendant la diastole réalisant
un phénomène du type « tonneau des
danaïdes ». Le recaptage du Ca2+ est sous la
dépendance d’un complexe macromoléculaire
organisé autour de la Ca2+-ATPase du RS
(SERCA2a) et de son régulatome c’est-à-dire
l’ensemble des protéines associées qui en régu-
lent à chaque instant le fonctionnement
(figure), parmi lesquelles le phospholamban
(PLN) qui exerce sur SERCA2a un rôle inhibiteur
quand il est déphosphorylé, les kinases qui le
phosphorylent, des phosphatases, des inhibi-
teurs de phosphatases, etc.
De même, du côté de la libération du Ca2+, les
canaux récepteurs de la ryanodine, RyR2, sont
aux centre d’un des plus importants complexes
macromoléculaires de l’organisme dont la
régulation est moins claire que celle de
SERCA2a et fait l’objet d’importantes contro-
verses.
Le troisième acteur principal des flux calciques
du myocyte est l’échangeur Na+-Ca2+ (NCX1)
qui fait sortir en diastole hors de la cellule le
Ca2+ qui y est entré pendant la systole par les
canaux calciques de type L pour déclencher la
libération de Ca2+ du RS via RyR2. Au-dessus
de ces « acteurs directs » des flux calciques du
myocyte il existe des sortes d’orchestrateurs
situés pour partie dans les régulatomes et qui
régulent à chaque instant le fonctionnement
de la principale protéine : SERCA2a, RyR2 et
NCX1. Parmi ces acteurs, la Ca2+ -calmoduline
kinase de type II (CaMKII) est maintenant
reconnue comme un des principaux respon-
Nouvelles cibles thérapeutiques
de l’insuffisance cardiaque
J.-J. Mercadier
Université Paris Diderot – Sorbonne Paris-Cité
Inserm UMRS 769 – LabEx LERMIT
Service de physiologie - explorations fonctionnelles, Groupe hospitalier Bichat – Claude-Bernard

PHYSIOPATHOLOGIE CARDIOVASCULAIRE
35AMC pratique n°229 juin 2014
J.-J. Mercadier
sables de la dégradation fonctionnelle du
myocyte cardiaque dans l’IC systolique
avancée. Au cours des dernières années,
chacun de ces grands complexes et parte-
naires a progressivement émergé en tant que
cible de grand intérêt potentiel pour le déve-
loppement de nouvelles thérapies de l’IC.
Quatre cibles majeures
SERCA2a
La sous-expression de SERCA2a observée dans
l’insuffisance cardiaque systolique représente
depuis sa découverte [1] la cible thérapeu-
tique privilégiée pour la thérapie génique
visant à rétablir un taux d’expression normal.
Roger Hajjar et son équipe sont les principaux
contributeurs du domaine avec des études de
phase II utilisant l’AAV1.SERCA2a (MYDICAR)
transféré aux myocytes cardiaques par injec-
tion intra-coronaire (étude CUPID) [2]. Un
problème parmi d’autres est que, au cours de
l’IC avancée, le peu de SERCA2a qui reste est
inhibé par un PLN déphosphorylé du fait
d’une activité excessive d’une phosphatase,
PP1, ce qui a fait de cette phosphatase une
cible thérapeutique à part entière. De fait, le
transfert par AAV (adeno-associated virus)
d’un inhibiteur de cette phosphatase préserve
la phosphorylation du PLN ce qui le découple
de SERCA2a, améliore la fonction contractile
et prévient le remodelage cardiaque à long
terme dans un modèle d’insuffisance cardiaque
par surcharge de pression chez le rat [3]. De
façon plus pharmacologique, un travail récent
vient de montrer que l’istaroxime, molécule
déjà testée dans l’IC aiguë et connue pour
Figure. Le myocyte cardiaque est représenté avec son réticulum sarcoplasmique (RS) et ses myofibrilles (MF, en noir en bas). Les
flèches noires représentent les mouvements du Ca2+.
A gauche, l’entrée de Ca2+ par les canaux Ca2+ de type L déclenche l’ouverture des récepteurs de la ryanodine (RyR) ce qui
permet au Ca2+ de sortir du RS pour aller déclencher la contraction au niveau des MF. Le Ca2+ est repompé dans le RS par
SERCA couplée à son inhibiteur, le phospholamban (PLN). Ce dernier peut être phosphorylé par une PKA (en rouge) et une CaMK
(en bleu) ce qui a pour effet de lever son effet inhibiteur sur SERCA qui repompe le Ca2+ plus vite (accélération de la relaxation
sous l’effet des catécholamines). RyR peut également être phosphorylé par une PKA et une CaMK ce qui favorise la sortie,
normale ou pathologique (voir détails dans le texte) de Ca2+ hors du RS. L’échangeur Na+-Ca2+ (NCX) éjecte le Ca2+ entré par
les canaux Ca2+ de type L hors de la cellule pour maintenir l’homéostasie cellulaire calcique. Il expulse 1 Ca2+ contre l’entrée
de 3 Na+ ce qui a un effet dépolarisant (flèches en pointillé rouge) qui, dans certaines situations pathologiques, est générateur
d’arythmies.

PHYSIOPATHOLOGIE CARDIOVASCULAIRE
Nouvelles cibles thérapeutiques de l’insuffisance cardiaque
36 AMC
pratique n°229 juin 2014
améliorer la relaxation, agit en stimulant
SERCA2a par un mécanisme analogue de
découplage entre l’enzyme et le PLN inhibi-
teur [4]. D’autres partenaires du régulatome
de SERCA2a ont été récemment identifiés
(HSP-20, HAX-1… [5]) parmi lesquels la
protéine SUMO1 semble constituer une cible
thérapeutique de choix. SUMO1 se comporte
comme une protéine chaperonne de SERCA2a
et la diminution de son expression dans l’IC
parallèle celle de SERCA2a et les altérations
fonctionnelles associées. Inversement, la
surexpression cardiaque de SUMO prévient le
remodelage cardiaque et normalise la survie
dans le modèle de la TAC chez la souris [6].
Toujours dans le domaine de la modulation
de l’expression ou de la fonction de SERCA2a,
une étude récente cible cette fois un micro-
ARN, miR-25, qui contrôle l’expression de
SERCA2a en déstabilisant son ARNm. Ainsi, le
transfert d’un micro-ARN antagoniste de
miR-25 (antagomir) prévient le remodelage
délétère et l’IC dans le modèle de la TAC chez
la souris [7].
Enfin, plutôt à classer comme un développe-
ment biotechnologique que comme l’identifi-
cation d’une nouvelle cible, l’équipe de
Joseph Metzger a montré dans une étude très
élégante que le transfert d’une parvalbu-
mine, protéine normalement pas exprimée
dans les myocytes cardiaques, mutée pour
diminuer la fixation des ions Ca2+ au profit des
ions Mg2+ crée un tampon calcique original
qui mime les effets de la stimulation adréner-
gique – augmentation de la contractilité et
accélération de la relaxation – sans en avoir
les inconvénients, notamment sur la consom-
mation d’oxygène du myocarde [8].
RyR2
La fuite du Ca2+ hors du RS pendant la diastole
représente également une cible thérapeu-
tique majeure. Bien que ceci reste débattu, il
existe des bases théoriques et expérimentales
importantes indiquant que cette fuite est au
moins en partie responsable de la dysfonction
systolique, de certaines arythmies et de la
stimulation des voies de signalisation du
remodelage délétère au cours des cardiopa-
thies évoluant vers l’IC. La controverse la plus
importante porte sur le rôle de la phospho-
rylation de la sérine 2808 de RyR2 par la PKA
tenue pour responsable de la dégradation
fonctionnelle et des arythmies par le groupe
d’Andrew Marks tandis que d’autres contes-
tent farouchement cette théorie [9, 10]. Le
rôle délétère de la phosphorylation par la
calcium-calmoduline kinase de type II b
(CaMKII b) de la sérine 2814 n’est, en revanche,
pas contesté. Cette sérine est hype rphospho-
rylée dans des modèles expérimentaux d’IC
et, chez l’homme, dans les cardiomyopathies
dilatées [11]. La prévention de cette phospho-
rylation en substituant cette sérine par une
alanine prévient le remodelage délétère dans
le modèle de la TAC chez la souris [11] et l’in-
hibition de la CaMKII par pré incubation avec
un peptide inhibiteur l’AIP fait disparaître les
fuites calciques observées sur des myocytes
cardiaques isolés de ventricules humains
défaillants [12].
De façon un peu surprenante, il semble que la
normalisation du taux de SERCA2a par trans-
fert d’ADN corrige également les anomalies
observées au niveau de RyR2 sans que le méca-
nisme de cette correction soit totalement
élucidé [13]. Enfin, les molécules dérivées de
la 1,4-benzothiazepine du type K201 (alias
JTV-519) préviennent la fuite diastolique de
Ca2+ hors du RS en favorisant la liaison entre
RyR2 et la protéine régulatrice FKBP12.6. Nous
avons montré que la prévention des arythmies
ventriculaires déclenchées par stimulation
ventriculaire rapide en présence d’une hypers-
timulation sympathique dans le modèle de la
TAC chez la souris par surexpression de
FKBP12.6 est associée à une moindre phospho-
rylation de la S2814 [14]. Notre hypothèse est
que la surexpression de FKBP12.6 protège la
S2814 de la phosphorylation par la CaMKII b.
NCX1
NCX1 est depuis longtemps une cible théra-
peutique potentielle pour la prévention des
arythmies déclenchées par la fuite de Ca2+
hors du RS. En effet, cette fuite active la sortie
d’1 Ca2+ contre l’entrée de 3 Na+ ce qui génère
un courant entrant dépolarisant responsable
des post-dépolarisations retardées généra-
trices d’arythmies. En revanche, l’effet du
blocage de NCX a été moins étudié en tant
que cible pour un traitement du déficit
contractile car on craint l’augmentation de la
concentration diastolique de Ca2+ et l’altéra-

PHYSIOPATHOLOGIE CARDIOVASCULAIRE
37AMC pratique n°229 juin 2014
J.-J. Mercadier
tion de la relaxation susceptible d’en décou-
ler. Ceci n’est pas observé dans tous les
modèles et il a été montré qu’un inhibiteur
de NCX, le SAE-0400 a un effet inotrope
positif dans le modèle de la TAC [15]. De plus,
cette molécule prévient la fibrose myocar-
dique dans un modèle d’HTA, probablement
du fait que NCX fonctionnant en mode
reverse (entrée de Ca2+ dans la cellule) stimule
la croissance et la différenciation des fibro-
blastes. Ceci pourrait conférer au SAE-0400 un
rôle thérapeutique spécifique dans l’IC à FE
préservée [16].
CaMKII
Des conséquences de l’hyperphosphorylation
de la sérine 2814 de RyR2 décrites plus haut et
de nombreuses autres altérations qui lui sont
liées, il ressort que CaMKII pourrait constituer
une cible majeure pour le traitement et surtout
la prévention de l’IC. Il est d’ailleurs admis que
bon nombre des effets bénéfiques des bêta-
bloqueurs dans l’IC, notamment leur effet anti-
arythmique, passe par une diminution de
l’activité de CaMKII. Le KO de la CaMKII b dans
le cœur de souris prévient le remodelage délé-
tère induit par la TAC [17, 18]. CaMKII b est
surexprimée dans les cœurs humains défaillants
et son inhibition par une pré incubation des
myocytes avec deux de ses inhibiteurs (AIP ou
KN-93) fait disparaitre les fuites calciques dias-
toliques. Il existe, dans les myocytes cardiaques,
plusieurs isoformes de CaMKII réparties diffé-
remment dans les divers compartiments cellu-
laires. Compte tenu d’effets potentiellement
délétères d’une inhibition globale de cette
enzyme centrale et ubiquitaire, le problème va
donc être de parvenir à inhiber une isoforme
donnée dans un compartiment donné pour
bloquer spécifiquement les effets délétères
ciblés.
Conclusion
Les quelques exemples décrits ci-dessus
montrent bien que l’on est entré dans une
nouvelle ère où les futures thérapies ne
consisteront plus seulement en de nouvelles
molécules chimiques douées d’effets pharma-
cologiques. Le transfert d’ADNc permettant
de compenser un déficit ou de réguler l’ex-
pression d’une protéine est en train d’entrer
dans la phase d’application clinique. De la
même manière, de nouvelles molécules
douées d’effets thérapeutiques telles que les
antagomirs feront un jour leur apparition en
cardiologie. Ces agents pourraient d’ailleurs
devenir un moyen très élégant et efficace
d’atteindre spécifiquement une cible intracel-
lulaire telle la CamKII b. Ces nouvelles inter-
ventions thérapeutiques seront mises en
œuvre de plus en plus tôt dans le processus
pathologique pour prévenir le remodelage
délétère. Dans les suites de l’infarctus du
myocarde, il faudra compter également avec
les moyens de stimuler le renouvellement des
myocytes détruits à partir de divers types
cellulaires.
Conflits d’intérêt : l’auteur déclare ne pas avoir de
conflits d’intérêt en relation avec cet article.
Références
[1] Mercadier JJ, Lompré AM, Duc P, et al. Altered sarcoplasmic
reticulum Ca2(+)-ATPase gene expression in the human
ventricle during end-stage heart failure. J Clin Invest
1990;85:305-9.
[2] Hajjar RJ. Potential of gene therapy as a treatment for heart
failure. J Clin Invest 2013;123:53-61.
[3] Pritchard TJ, Kawase Y, Haghighi K, et al. Active inhibitor-1
maintains protein hyper-phosphorylation in aging hearts and
halts remodeling in failing hearts. PLoS One 2013:e80717.
doi: 10.1371/journal.pone.0080717. eCollection 2013.
[4] Ferrandi M, Barassi P, Tadini-Buoninsegni F, et al. Istaroxime
stimulates SERCA2a and accelerates calcium cycling in heart
failure by relieving phospholamban inhibition. Br J Pharmacol
2013;169:1849-61. doi: 10.1111/bph.12278.
[5] Kranias EG, Hajjar RJ. Modulation of cardiac contractility by
the phospholamban/SERCA2a regulatome. Circ Res
2012;110:1646-60. doi: 10.1161/CIRCRESAHA.111.259754.
Review.
[6] Kho C, Lee A, Jeong D, et al. SUMO1-dependent modulation
of SERCA2a in heart failure. Nature 2011;477:601-5.
[7] Wahlquist C, Jeong D, Rojas-Muñoz A, et al. Inhibition of
miR-25 improves cardiac contractility in the failing heart.
Nature 2014;508:531-5.
[8] Wang W, Barnabei MS, Asp ML, et al. Noncanonical EF-hand
motif strategically delays Ca2+ buffering to enhance cardiac
performance. Nat Med 2013;19:305-12.
[9] Houser SR. Role of RyR2 Phosphorylation in heart failure and
arrhythmias: Protein kinase A-mediated hyperphosphorylation
of the ryanodine receptor at serine 2808 does not alter car-
diac contractility or cause heart failure and arrhythmias. Circ
Res 2014;114:1320-7.

PHYSIOPATHOLOGIE CARDIOVASCULAIRE
Nouvelles cibles thérapeutiques de l’insuffisance cardiaque
38 AMC
pratique n°229 juin 2014
[10] Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart
failure and arrhythmias: controversies around ryanodine
receptor phosphorylation in cardiac disease. Circ Res
2014;114:1311-9.
[11] Respress JL, van Oort RJ, Li N, et al. Role of RyR2 phosphory-
lation at S2814 during heart failure progression. Circ Res
2012;110:1474-83.
[12] Fischer TH, Herting J, Tirilomis T, et al. Ca2+/calmodulin-
dependent protein kinase II and protein kinase A differentially
regulate sarcoplasmic reticulum Ca2+ leak in human cardiac
pathology. Circulation 2013;128:970-81.
[13] Lyon AR, Bannister ML, Collins T, et al. SERCA2a gene transfer
decreases sarcoplasmic reticulum calcium leak and reduces
ventricular arrhythmias in a model of chronic heart failure. Circ
Arrhythm Electrophysiol 2011;4:362-72.
[14] Vinet L, Pezet M, Bito V, et al. Cardiac FKBP12.6 overexpres-
sion protects against triggered ventricular tachycardia in
wressure overloaded mouse hearts. Basic Res Cardiol
2012;107:246.
[15] Antoons G, Willems R, Sipido KR. Alternative strategies in
arrhythmia therapy: evaluation of Na/Ca exchange as an anti-
arrhythmic target. Pharmacol Ther 2012;134:26-42.
[16] Kamimura D, Ohtani T, Sakata Y, et al. Ca2+ entry mode of
Na+/Ca2+ exchanger as a new therapeutic target for heart
failure with preserved ejection fraction. Eur Heart J
2012;33:1408-16.
[17] Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase
II is required for pathological cardiac hypertrophy and remod-
eling after pressure overload. Proc Natl Acad Sci USA
2009;106:2342-7.
[18] Ling H, Zhang T, Pereira L, et al. Requirement for Ca2+/
calmodulin-dependent kinase II in the transition from pressure
overload-induced cardiac hypertrophy to heart failure in mice.
J Clin Invest 2009;119:1230-40.
1
/
5
100%