Lire l'article complet

Dossier
Act. Méd. Int. - Métabolismes - Hormones - Nutrition, Volume II, n° 4, août 1998
hypogonadisme hypogonadotro-
phique est défini par une diminution
de la synthèse des gonadotrophines FSH et
LH (1). L’origine de ce déficit est hypotha-
lamique ou hypophysaire. La majorité des
déficits gonadotropes est due à une patholo-
gie tumorale ou infiltrative. Outre ces défi-
cits acquis, il est décrit des déficits congéni-
taux (tableau I, page 6). Ceux-ci peuvent
être associés à une pathologie bien caracté-
risée, telle que l’hypoplasie congénitale des
surrénales, ou survenir isolément. Dans les
déficits isolés, deux grands groupes sont
décrits : le syndrome de Kallmann, défini
par un déficit gonadotrope associé à une
anosmie, et l’hypogonadisme hypogonado-
trophique dit idiopathique (HHI), compre-
nant les individus n’ayant pas d’anosmie.
Le gène du syndrome de Kallmann, lié au
chromosome X, a été cloné par clonage
positionnel. Des mutations ont ainsi pu être
décrites. Ces travaux ont confirmé que l’hé-
térogénéité clinique observée dans l’hypo-
gonadisme hypogonadotrophique était bien
due à une hétérogénéité génétique. Dans les
HHI, plusieurs groupes ont recherché des
anomalies du gène de la GnRH, sans suc-
cès. Le gène du récepteur de la GnRH était
un autre gène candidat logique. Cette hypo-
thèse a été testée dans une population de
patients présentant un déficit gonadotrope
isolé. Cela a permis la caractérisation de
plusieurs mutations inactivatrices du gène
du récepteur de la GnRH.
Les hypogonadismes hypogona-
dotrophiques congénitaux
Le syndrome de Kallmann
Le syndrome de Kallmann a été décrit pour
la première fois en 1856 par Maestre de San
Juan. En 1944, Kallmann publie le premier
cas familial. En 1954, de Morsier suggère
que l’anosmie pourrait être due à l’agénésie
des bulbes olfactifs. Ce syndrome est alors
nommé dysplasie olfacto-génitale ou syn-
drome de Kallmann-de Morsier. Des travaux
Mutations inactivatrices
du récepteur de la GnRH :
une nouvelle étiologie d’hypo-
gonadisme hypogonadotrophique
N. de Roux*, M. Misrahi*, J. Young**, G. Schaison**, E. Milgrom*
5
✎
L’hypogonadisme hypogonado-
trophique est un syndrome hétéro-
gène défini par une diminution de
la biosynthèse des hormones glyco-
protéiques FSH et LH.
✎
L
es déficits gonadotropes sont soit
congénitaux, soit acquis. Les déficits
congénitaux sont parfois associés à
une pathologie bien caractérisée, ou
surviennent isolément. Des études de
ségrégation ont montré l’origine
génétique des déficits gonadotropes
congénitaux isolés.
✎
Le syndrome de Kallmann asso-
cie un hypogonadisme hypogona-
dotrophique et une anosmie. Le
déficit gonadotrope survenant sans
anosmie définit l’hypogonadisme
hypogonadotrophique idiopathique.
✎
Le gène responsable de la forme
liée à l’X du syndrome de Kallmann
est cloné. Des mutations ont été
décrites. Le gène du récepteur de la
GnRH était un gène candidat de l’hy-
pogonadisme hypogonadotrophique
sans anosmie.
✎
Plusieurs mutations du gène du
récepteur de la GnRH ont été carac-
térisées dans des cas familiaux
d’hypogonadisme hypogonadotro-
phique. Les phénotypes varient
entre un déficit gonadotrope com-
plet et un déficit partiel. Ces muta-
tions altèrent la fonction du récep-
teur en diminuant la liaison de
l’hormone sur son récepteur ou la
transduction du signal.
✎
Une mutation inactivatrice est
donc une nouvelle étiologie de
l’hypogonadisme hypogonadotro-
phique sans anosmie.
* INSERM U 135 et laboratoire d’hormonologie et de biologie moléculaire,
** Service d’endocrinologie et des maladies de la reproduction, hôpital de Bicêtre,
94270 Le Kremlin-Bicêtre.
L’

Act. Méd. Int. - Métabolismes - Hormones - Nutrition, Volume II, n° 4, août 1998
ultérieurs, sur des coupes de cerveau d’un
fœtus atteint d’un syndrome de Kallmann,
montreront une absence de migration de la
placode olfactive vers l’hypothalamus des
neurones synthétisant la GnRH.
La transmission est préférentiellement liée
à l’X. Des transmissions autosomiques
récessives et dominantes ont également été
décrites. Le sex-ratio garçon-fille est de 7.
La fréquence du syndrome de Kallmann
dans la population générale est d’environ
1/10 000 (2).
L’expression clinique de ce syndrome est
variable. Dans le sexe masculin, le dia-
gnostic est évoqué devant un micropénis,
souvent associé à une cryptorchidie dans
l’enfance, un retard pubertaire dans l’ado-
lescence ou un hypogonadisme à l’âge
adulte. Dans le sexe féminin, le phénotype
va de l’impubérisme complet à une inferti-
lité par anovulation chronique.
Deux approches par génétique inverse ont
permis la localisation d’un gène en Xq23.1,
puis son clonage. Baptisé KAL, il comprend
14 exons et code pour une protéine de 680
acides aminés. La protéine KAL possède des
homologies de structure avec les protéines
d’adhésion des neurones. Un pseudogène est
localisé sur le bras long du chromosome Y,
en position Yq11, dans la région connue pour
être homologue au chromosome X (2).
Les études in vitro ont montré que la pro-
téine KAL est localisée à la surface des cel-
lules. Le mécanisme de cette localisation
est inconnu, puisque la protéine KAL ne
contient ni domaine transmembranaire ni
séquence consensus permettant l’accro-
chage d’une glycosylphosphatidylinositol
(GPI) pour ancrer la protéine à la mem-
brane plasmique. Par RT-PCR, il a été
montré que le gène KAL est exprimé dans
de nombreux tissus tels que le cerveau, le
foie, le rein, le muscle strié... L’altération
de la fonction de la protéine KAL dans ces
tissus pourrait participer à la survenue de
l’agénésie rénale unilatérale ou à celle des
signes neurologiques parfois observés dans
le syndrome de Kallmann.
La confirmation du rôle du gène KAL a été
possible grâce à la caractérisation de délé-
tions de la séquence codante et à la des-
cription de mutations ponctuelles. Surtout
des mutations non-sens entraînant un arrêt
du cadre de lecture et la synthèse d’une
protéine tronquée ont définitivement
confirmé le rôle de la protéine KAL. Les
relations génotype-phénotype sont mal
comprises. Une variabilité de l’expression
phénotypique a notamment été observée
dans plusieurs familles (2).
Finalement, ces travaux ont confirmé que
l’hétérogénéité du tableau clinique observé
chez les patients ayant un hypogonadisme
hypogonadotrophique était bien due à une
hétérogénéité génétique. En effet, les ano-
malies du gène KAL concernent moins de
15 % de tous les hypogonadismes hypogo-
nadotrophiques avec ou sans anosmie. Il
fallait donc rechercher d’autres gènes can-
didats. La seule anomalie connue était une
translocation entre les chromosomes 7 et
12 dans la forme dominante.
L’hypogonadisme hypogonado-
trophique dit idiopathique (HHI)
L’hypogonadisme hypogonadotrophique
dit idiopathique (HHI) regroupe les
patients n’ayant pas d’anosmie ou les stig-
mates cliniques parfois retrouvés dans le
syndrome de Kallmann. Les formes fami-
liales représentent une faible proportion de
cette population, composée essentiellement
de cas sporadiques. Un mode de transmis-
sion autosomique récessif a été le plus sou-
vent proposé. La présentation clinique de
l’hypogonadisme ne diffère pas de celle
retrouvée dans le syndrome de Kallmann.
En effet, il existe des déficits complets et
des formes partielles équivalentes au
tableau clinique des eunuques fertiles.
L’étude de la fréquence et de l’amplitude
des pics de sécrétion de la LH au cours du
temps permet de distinguer la forme com-
plète de la forme partielle. Les patients
ayant un déficit complet n’ont pas de pic de
sécrétion de la LH, aussi bien diurne que
nocturne. En revanche, dans les déficits
incomplets, la fréquence ou l’amplitude
des pics de sécrétion sont diminuées mais
pas abolies.
L’hypogonadisme hypogonadotrophique
idiopathique a été comparé au déficit gona-
dotrope survenant chez la souris hypogona-
dique (hpg). La GnRH est indétectable dans
l’hypothalamus de ces souris, alors que
l’hypophyse répond normalement à une sti-
mulation par de la GnRH exogène. Il était
donc logique de rechercher une anomalie
du gène de la GnRH. Mason et coll. ont
démontré, en 1986, que ce déficit était dû à
une délétion d’au moins 33 Kpb compre-
nant les exons 3 et 4 du gène de la pro-hor-
mone GnRH-GAP (3). Ces deux exons
codent pour la partie C-terminale de la pro-
téine GAP (GnRH associated protein), alors
que le décapeptide GnRH est codé par
l’exon 2. Le gène délété étant normalement
transcrit dans l’hypothalamus, il est pro-
bable que l’intégrité structurale de la pro-
hormone GnRH-GAP soit indispensable à
la maturation post-traductionnelle. La réin-
troduction par thérapie génique de ce gène
6
Dossier
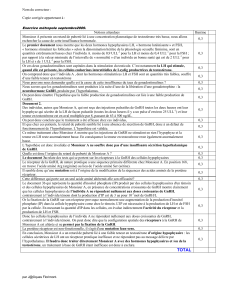
Tableau I. Étiologies des hypogonadismes hypo-
gonadotrophiques.
Hypogonadismes hypogonadotrophiques
acquis :
●Tumeurs : craniopharyngiome, adénomes
hypophysaires ...
●Hyperprolactinémie.
●Syndrome de Sheehan.
●Hémochromatose.
●Sarcoïdose.
●Radiothérapie.
●Anorexie mentale.
Hypogonadismes hypogonadotropiques
congénitaux :
●Avec anosmie :
– Syndrome de Kallmann.
●Sans anosmie :
– Mutations inactivatrices
du récepteur de la GnRH.
– Mutations inactivatrices de la LH.
– Mutations inactivatrices de la FSH.
– Hypoplasie congénitale
des surrénales.
– Mutations de la leptine
ou de son récepteur.

Act. Méd. Int. - Métabolismes - Hormones - Nutrition, Volume II, n° 4, août 1998
a permis une restauration complète des
fonctions de reproduction de la souris hpg.
Néanmoins, aucune anomalie du gène de
GnRH n’a été retrouvée chez l’homme,
aussi bien par Southern-Blot, afin de
rechercher des anomalies majeures du gène,
que par séquençage direct de la séquence
codante.
Les hypogonadismes liés à une
anomalie de la LH ou de la FSH
Parfois, l’anomalie concerne une des deux
gonadotrophines. Les déficits gonado-
tropes dus à un déficit isolé de la LH ou de
la FSH sont très rares. Une mutation homo-
zygote (Gln54Arg) de la sous-unité ß de la
LH a été trouvée chez un homme ayant un
retard pubertaire. Cette mutation inhibait
complètement la liaison de la LH à son
récepteur (4). Deux cas de déficit isolé en
FSH dus à des mutations inactivatrices de
la sous-unité ß de la FSH ont été décrits
chez deux femmes ayant une aménorrhée
primaire (5, 6). Une patiente était homozy-
gote pour une délétion de deux nucléotides
(codon 61) et l’autre hétérozygote compo-
site pour la même délétion et une substitu-
tion de la cystéine 51 en une glycine. La
délétion de deux nucléotides au codon 61
est responsable de la synthèse d’une protéi-
ne tronquée. Ces deux mutations diminuent
la bioactivité de la FSH. La même délétion
de deux nucléotides du gène de la FSH
vient d’être décrite à l’état homozygote
chez un homme ayant un hypogonadisme
et une azoospermie (7).
Les déficits gonadotropes asso-
ciés à une autre pathologie
congénitale
Un hypogonadisme hypogonadotrophique
est décrit dans l’hypoplasie congénitale des
surrénales liée à une mutation de DAX-1.
L’obésité due à des mutations de la leptine
ou de son récepteur est également associée
à un hypogonadisme hypogonadotro-
phique. Les mécanismes de ces déficits
gonadotropes restent incompris. Un déficit
gonadotrope a également été décrit chez un
patient ayant un déficit global de la matu-
ration des pro-hormones hypophysaires.
Un gène candidat:
le récepteur de la GnRH
Un déficit endocrinien peut être dû à un
déficit de la synthèse ou de la bioactivité de
l’hormone, mais également à un déficit
fonctionnel de son récepteur. Des muta-
tions inactivatrices de plusieurs récepteurs
couplés aux protéines G ont été caractéri-
sées dans diverses pathologies endocri-
niennes (voir MHN vol. I, n° 1. Mutations
du récepteur de la TSH et hyperthyroïdie,
p. 8-14). Néanmoins, le gène du récepteur
de la GnRH avait été réfuté par de nom-
breux auteurs en tant que gène candidat à
l’hypogonadisme hypogonadotrophique
sans anosmie. L’argument majeur étant la
réponse positive à une injection unique de
GnRH observée chez les plupart des
patients. Cette interprétation ne tenait pas
compte des possibilités d’une atteinte par-
tielle de ce récepteur, qui serait invisible
lors de l’injection d’une dose pharmacolo-
gique de GnRH (100 µg i.v.). En effet,
notre expérience acquise dans l’étude des
mutations inactivatrices du récepteur de la
TSH et de la LH avait montré que le phé-
notype de ces mutations pouvait varier
entre un déficit complet et un déficit par-
tiel. Nous avons donc recherché des ano-
malies du gène du récepteur de la GnRH
dans plusieurs cas familiaux d’hypogona-
disme hypogonadotrophique partiel ou
complet. Ceci nous a permis de décrire le
premier cas de déficit gonadotrope par
mutations inactivatrices du récepteur de la
GnRH (8).
Le clonage de l’ADNc du récepteur de la
GnRH a confirmé que ce récepteur appar-
tient à la super-famille des récepteurs cou-
plés aux protéines G (9). En effet, il est
formé de 328 acides aminés chez l’homme
et de 327 acides aminés chez la souris et le
rat. La protéine comprend sept domaines
transmembranaires, reliés par des boucles
intracellulaires et extracellulaires, et un
domaine extracellulaire. L’architecture de
ce récepteur est identique à celle d’autres
récepteurs couplés aux protéines G, sauf
sur un point. Tous les récepteurs de la
GnRH clonés chez les mammifères ne pos-
sèdent pas de domaine intracellulaire.
Deux sites de N-glycosylation sont décrits
dans le domaine extracellulaire et dans la
première boucle extracellulaire du récep-
teur humain. La séquence primaire en
acides aminés est très conservée parmi les
espèces, mais l’homologie avec les autres
RCPG est faible. Le gène du récepteur de
la GnRH comprend trois exons. Ce gène,
localisé sur le bras long du chromosome 4,
est unique (9).
Dans l’hypophyse humaine, trois bandes
sont visibles en Northern-Blot : une bande
majoritaire de 4,5 kb et deux bandes mino-
ritaires de 2,5 et 1,5 kb. La différence struc-
turale entre ces trois bandes est actuelle-
ment inconnue. Elle résulte certainement
d’un épissage alternatif de l’ARN pré-mes-
sager. Deux variants de l’ARNm du récep-
teur de la GnRH sont décrits chez la souris
et chez l’homme. Un de ces variants ne
contient pas le deuxième exon. Il code pour
une protéine de 177 acides aminés. Le
deuxième variant est obtenu par l’utilisation
d’un autre site accepteur d’épissage, locali-
sé dans le deuxième exon. Il code pour une
protéine tronquée de 249 acides aminés.
Dans des cellules humaines 293, cette pro-
téine tronquée a un effet inhibiteur sur la
stimulation de la phospholipase C par la
forme complète du récepteur de la GnRH.
L’ARNm du récepteur de la GnRH a été
mis en évidence par RT-PCR dans l’ovaire,
le sein, le testicule, la prostate et le placen-
ta. La finalité physiologique de ces locali-
sations est encore inconnue (9).
Les voies de transduction du signal par le
récepteur de la GnRH sont complexes. La
voie principale est la stimulation de la
phospholipase Cß par l’intermédiaire des
protéines Gq/G11. Il en résulte une synthè-
se d’IP3 et de diacylglycérol responsables
7

Act. Méd. Int. - Métabolismes - Hormones - Nutrition, Volume II, n° 4, août 1998
respectivement de la mobilisation du cal-
cium intracellulaire et de l’activation de la
protéine kinase C. La phospholipase D et la
phospholipase A2 sont également activées
par la GnRH. L’augmentation du calcium
intracellulaire est suivie par une entrée de
calcium extracellulaire par l’intermédiaire
de canaux voltage-dépendants. Récemment,
il a été montré que le récepteur de la GnRH
régulait l’activité des MAP-kinases par
l’intermédiaire des protéines kinases C.
Les MAP-kinases participent à la transduc-
tion du signal de nombreux récepteurs tels
que les récepteurs de facteur de croissance
ou d’autres récepteurs couplés aux pro-
téines G.
Les mécanismes de régulation de la trans-
cription des gènes des sous-unités αet ß de
la LH et de la FSH diffèrent. La transcrip-
tion du gène de la sous-unité αest stimulée
par administrations pulsatiles ou continues
de GnRH. En revanche, celle du gène de la
sous-unité ß n’est possible que par les
administrations pulsatiles de GnRH. Ces
différences pourraient être dues à l’utilisa-
tion de voies de transduction différentes
régulant l’expression des gènes des sous-
unités αet ß : la voie de la MAP-kinase
pour la sous-unité αet l’augmentation du
calcium intracellulaire par l’intermédiaire
des canaux Ca++ pour la sous-unité ß.
Les mutations naturelles
inactivatrices du récepteur
de la GnRH
Les trois exons du gène du récepteur de la
GnRH ont été séquencés chez plusieurs
patients. Deux mutations ont été retrouvées
dans une famille multiplexe (8). Le propo-
situs était un garçon ayant consulté pour
hypogonadisme. Les explorations hormo-
nales statiques et dynamiques ont montré
que ce déficit gonadotrope était partiel. Le
test à la GnRH était notamment positif.
L’amplitude des pics de sécrétion sponta-
née de la LH était diminuée (figure 1) mais
pas nulle. Une sœur du propositus avait des
antécédents d’aménorrhée primaire et d’in-
fertilité, malgré un développement puber-
taire normal. L’œstradiol plasmatique, les
concentrations de la FSH, de la LH et le
test à la GnRH étaient normaux. En
revanche, le volume ovarien était diminué
sans follicule de plus de 10 mm.
Deux mutations du
récepteur de la GnRH
ont été trouvées chez le
propositus et sa sœur.
L’étude familiale a
montré qu’ils étaient
tous les deux hétérozy-
gotes composites (une
mutation hétérozygote
sur chaque allèle). Ces
données concordaient
avec la transmission
autosomique récessive
habituellement obser-
vée dans les pathologies
dues à des mutations
inactivatrices des récep-
teurs couplés aux pro-
téines G. Une mutation
changeait la glutamine
106, localisée dans la
première boucle extra-
cellulaire, en une argini-
ne. Cette mutation
diminuait considérable-
ment la liaison de la
GnRH à son récepteur
(figure 2). La deuxième
mutation transformait
l’arginine 262 en une
glutamine. Cette derniè-
re, localisée dans la troi-
sième boucle intracellu-
laire, ne modifiait pas la
liaison de la GnRH
mais diminuait la trans-
duction du signal (8).
Depuis la description
de ce premier cas, nous
avons caractérisé une
nouvelle mutation dans
une autre famille comprenant trois sujets
malades. Cinq familles (13 individus
atteints) sont maintenant décrites (trois
françaises, une américaine et une suisse)
(10, 11, 12). La comparaison des phéno-
types de ces cinq familles semble montrer
que le tableau clinique de l’hypogonadisme
par perte de fonction du récepteur de la
8
Dossier
Figure 1. Étude de la pulsabilité de la sécrétion spontanée de la LH.
A. Étude chez un garçon ayant un déficit gonadotrope par mutation
inactivatrice du récepteur de la GnRH.
B. Sujet normal. T : testostérone plasmatique.

Act. Méd. Int. - Métabolismes - Hormones - Nutrition, Volume II, n° 4, août 1998
GnRH est très divers. En effet, la profon-
deur de l’atteinte de la fonction gonadotro-
pe est variable entre individus non apparen-
tés, mais également dans une même famil-
le. Les variations phénotypiques entre les
sujets malades pourraient être en rapport
avec l’anomalie moléculaire. Plus une
mutation altère la fonction du récepteur in
vitro, plus le déficit gonadotrope serait pro-
fond. Néanmoins, cette relation phénotype-
génotype est probablement perturbée par
d’autres facteurs. En effet, les variations
intrafamiliales observées dans certains cas
suggèrent qu’il existe des mécanismes de
compensation de l’altération de la fonction
gonadotrope. Ces mécanismes pourraient
dépendre du patrimoine génétique de
chaque sexe ou de chaque individu, voire de
facteurs épigénétiques.
La mutation Arg262Gln est retrouvée dans
quatre cas sur cinq (figure 3). Il n’existe
pas de lien de parenté entre les sujets por-
teurs de cette mutation. Il semble donc
exister un “point chaud” de mutations dans
la troisième boucle intracellulaire.
Un nombre plus important de cas doit être
décrit pour confirmer cette observation.
La recherche de mutations naturelles res-
ponsables de la survenue d’une pathologie
apporte de nombreuses informations sur la
relation structure-fonction des protéines.
Ainsi, dans le cadre du récepteur de la
GnRH, les mutations décrites permettent de
définir les régions participant à la liaison du
ligand et celles participant à la transduction
du signal. Des études in vitro ont montré le
rôle joué par la troisième boucle intracellu-
laire des récepteurs couplés aux protéines G
dans la transduction du signal. La caractéri-
sation de la mutation Arg262Gln confirme
ces observations. De même, il a été montré,
dans d’autres récepteurs couplés aux pro-
téines G, le rôle important joué par les
domaines transmembranaires et les boucles
extracellulaires dans la liaison du ligand.
Nos résultats sont en faveur d’un mécanis-
me similaire pour le récepteur de la GnRH.
9
Figure 2. Étude fonctionnelle des mutations
Gln106Arg et Arg262Gln. Les deux récepteurs
mutés sont exprimés dans des cellules hétéro-
logues in vitro.
A. Étude de la liaison de la GnRH sur le
récepteur (”100 %” correspond à la liaison
maximale observée pour le récepteur nor-
mal).
B. Étude de l’accumulation des inositols
phosphates (”100 %” correspond à la sti-
mulation maximale pour le récepteur nor-
mal).
Figure 3. Représentation schématique du récepteur de la GnRH avec la localisation de ses mutations natu-
relles inactivatrices.
* : nombre de cas décrits.
 6
6
1
/
6
100%