L`HYPOGONADISME HYPOGONADOTROPE

ENDOCRINOLOGIE
8 ADOLESCENCE & Médecine • Juin 2016 • numéro 11
L’HYPOGONADISME
HYPOGONADOTROPE CONGÉNITAL
CHEZ L’ADOLESCENTE
Un diagnostic facile?
La connaissance des gènes en cause pour les hypogonadismes hypo-
gonadotropes est en pleine progression depuis une dizaine d’années.
Ces identifications permettent d’améliorer le conseil génétique et la
prise en charge précoce des patients, y compris dans les formes diciles.
Exemple avec un cas clinique.
CAS CLINIQUE
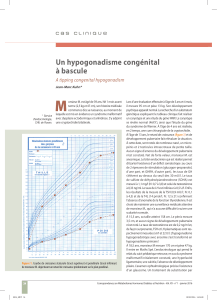
Armelle consulte à l’âge de 15 ans pour
un retard statural à - 2,5 DS et un retard
pubertaire (visible sur sa courbe de
croissance). Ses parents mesurent 159 et
165 cm. Les paramètres de naissance
sont normaux (TN 49 cm). Armelle est
A1P2S1R0. Elle mesure 148 cm et pèse
41 kg, elle est scolarisée en seconde. Son
examen clinique est sans particularité
en dehors de l’impubérisme et elle a un
odorat normal. L’analyse de la courbe de
croissance montre un infléchissement
de la vitesse de croissance dès l’âge de
11 ans. Un bilan général, hypophysaire
et osseux, est prescrit.
La NFS est normale (Hb 13 g/dl), la
VS est mesurée à 21 mm. La T4L est à
19 pmol/l, l’ACTH à 48 pg/ml, le corti-
sol à 500 nmol/l et l’IGF1 à 380 ng/ml.
La FSH est mesurée à 1,09 UI/l et la LH
à 0,2 UI/l. Le dosage du cortisol libre
urinaire sur 24 heures est à 32 µg/24 h.
L’âge osseux est à 12 ans. Une IRM céré-
brale et hypophysaire est prescrite rapi-
dement, elle est normale, les bulbes ol-
factifs sont vus. En raison du syndrome
inflammatoire, vu l’âge de la patiente et
malgré son IMC à 22 et l’absence totale
de signes fonctionnels abdominaux,
une coloscopie est réalisée avec des
biopsies : une maladie inflammatoire
du tube digestif est éliminée. La VS sera
recontrôlée secondairement à 13 mm.
À l’âge de 15 ans et demi, un traitement
par 17 bêta-estradiol puis œstroproges-
tatifs est débuté. Il permet un dévelop-
pement pubertaire complet et la surve-
nue de règles. La taille finale est 155 cm
(taille cible). En l’absence de diagnostic
étiologique, l’ADN de la patiente a été
testé sur un panel de gènes d’hypogo-
nadisme hypogonadotrope. Elle est
porteuse d’une mutation hétérozygote
pathogène connue de FGFR1. Armelle
est donc atteinte d’un syndrome de
Kallmann par mutation de FGFR1.
L’HYPOGONADISME
D’ORIGINE HYPOTHALAMO-
HYPOPHYSAIRE
L’hypogonadisme d’origine hypothala-
mo-hypophysaire, ou hypogonadisme
hypogonadotrope (HH), est défini par
une production ou une action insuffi-
sante de la GnRH et/ou des gonadotro-
phines. Le déficit est isolé ou combiné
lorsqu’il est associé à un autre déficit hy-
pophysaire (somatotrope, thyréotrope
ou corticotrope). Un hypogonadisme
hypogonadotrope isolé ou combiné
est syndromique lorsqu’il est associé à
des signes cliniques non endocriniens.
Les formes congénitales d’HH sont
classées de façon historique en deux
groupes, avec (syndrome de Kallmann
de Morsier) et sans anosmie. Mais cette
classification est probablement très
artificielle. Les HH congénitaux restent
des pathologies très rares : leur pré-
valence chez la femme est estimée à
0,01 %.
D’abord, tout hypogonadisme hypogo-
nadotrope doit faire évoquer un pro-
cessus expansif hypothalamo-hypo-
physaire (craniopharyngiomes…) ou
inflammatoire (sarcoïdose, histiocytose,
hypophysites lymphocytaires). En pra-
tique, les causes d’infiltration de la tige
s’intègrent le plus souvent dans une
pathologie globale hypophysaire, avec
un diabète insipide. Elles sont excep-
tionnellement responsables d’un retard
pubertaire isolé. Dans tous les cas, avant
d’évoquer le diagnostic d’HH congéni-
tal, il est indispensable d’éliminer une
pathologie hypothalamo-hypophysaire
organique. Une fois ce diagnostic éli-
miné, il est difficile, devant l’absence
de développement pubertaire ou une
puberté peu évolutive chez une adoles-
cente, de distinguer un retard pubertaire
simple, même s’il est plus rare chez les
filles, d’un HH.
Le déficit gonadotrope isolé ne donne
pas de signe clinique chez les nouveau-
nés de sexe féminin, contrairement aux
garçons (micropénis, cryptorchidie) et
un HH sera donc évoqué devant l’ab-
sence de développement ou un déve-
loppement incomplet mammaire, une
Dr Claire Bouvattier
Endocrinologie pédiatrique,
hôpital Bicêtre, Centre de réfé-
rence des maladies du déve-
loppement génital, Faculté de
médecine Paris Sud

ENDOCRINOLOGIE
10 ADOLESCENCE & Médecine • Juin 2016 • numéro 11
aménorrhée primaire, plus rarement
une spanioménorrhée. L’interrogatoire
familial questionnera l’âge de la puberté
chez la mère et les sœurs. La courbe de
la croissance sera reconstituée : dans
les retards pubertaires simples, il existe
habituellement un ralentissement de la
vitesse de croissance. Les explorations
biologiques confirmeront l’HH : une
concentration d’estradiol basse, asso-
ciée à des concentrations basses ou pa-
radoxalement normales de FSH et LH.
La prolactine est normale.
Plus de 25 gènes responsables d’HH
congénitaux avec ou sans anosmie sont
connus à ce jour et un diagnostic géné-
tique est posé chez près de la moitié des
patients.
LE SYNDROME DE
KALLMANN
Le syndrome de Kallmann atteint
1/10 000 garçons et est 5 à 7 fois moins
fréquent chez les filles. L’HH est secon-
daire à un défaut de migration des
neurones à GnRH et le déficit olfactif
est imagé par une hypoplasie ou une
absence des bulbes olfactifs à l’IRM.
La plupart des cas sont diagnostiqués
devant l’absence de puberté spontanée,
associée à l’atteinte de l’odorat, mais
d’autres signes peuvent être associés :
agénésie rénale, syncinésies d’imitation,
troubles de l’oculomotricité, ptosis, syn-
drome cérébelleux, surdité, pieds creux,
fente labiale et/ou palatine...
Trois modes de transmission ont été dé-
crits dans les syndromes de Kallmann :
- une forme liée à l’X,
- une forme autosomique dominante,
- une forme autosomique récessive.
La forme de la maladie liée à l’X ne
s’exprime que chez le garçon. Le gène
FGFR1 (fibroblast growth factor recep-
tor 1) code pour le récepteur d’un fac-
teur de croissance qui agirait en étroite
liaison avec l’anosmine, dans la mise en
place de l’axe gonadotrope. Les muta-
tions identifiées chez les patients atteints
d’un syndrome de Kallmann avec une
transmission autosomique dominante
sont des mutations “perte de fonction”
qui expliqueraient 10 % des syndromes
de Kallmann. Récemment, de nouveaux
gènes ont été identifiés dans des syn-
dromes de Kallmann de transmission
dominante : FGF8, HS6ST1, SOX10,
SEMA3A, WDR11 et IL17RD.
Le syndrome CHARGE (Coloboma,
Heart disease, Choanal atresia,
Retarded growth and developpement,
Genital hypoplasia, Ear abnormali-
ties) est un syndrome polymalformatif
dont l’incidence est estimée autour de
1 pour 10 000 naissances. Le tableau
clinique comprend un hypogonadisme
hypogonadotrope avec une anosmie
et des anomalies des bulbes olfactifs
à l’IRM. Un des gènes mis en cause
dans le syndrome CHARGE est le gène
CHD7 qui code pour une protéine
impliquée dans la structure de la chro-
matine. Dans les HH avec anosmie, les
mutations de ProK2 sont retrouvées à
l’état hétérozygote, celles de ProKR2
à l’état homozygote ou hétérozygote.
L’expressivité phénotypique de ces
mutations est très variable. Des muta-
tions hétérozygotes (transmission au-
tosomique dominante) du gène NELF
(Nasal Embryonic LHRH Factor) ont
également été observées chez de rares
patients.
Figure 1 - Courbe de croissance du cas clinique.

L’hypogonadisme hypogonadotrope congénital chez l’adolescente
ADOLESCENCE & Médecine • Juin 2016 • numéro 11 11
LES HYPOGONADISMES
HYPOGONADOTROPES
ISOLÉS SANS ANOSMIE
Les premières mutations “perte de
fonction” du récepteur de la GnRH ont
été décrites en 1997 chez un homme
de 22 ans et sa sœur qui présentaient
un hypogonadisme hypogonadotrope
partiel. Cliniquement, le jeune homme
avait eu une puberté tardive et incom-
plète, et se plaignait de troubles de la
libido. Sa sœur aînée, âgée de 37 ans
présentait une aménorrhée primaire et
une infertilité. Le séquençage du gène
du récepteur de la GnRH a permis de
démontrer que ces deux patients étaient
hétérozygotes composites.
Depuis, d’autres mutations du récepteur
de la GnRH ont été décrites, qui altèrent
la liaison de la GnRH à son récepteur et/
ou la transduction du signal. Leur trans-
mission est autosomique récessive avec
des phénotypes variés.
Les familles porteuses d’une mutation
“perte de fonction” du gène codant pour
KISS1R ou son ligand KISS1 présentent
une absence de puberté liée à un déficit
de sécrétion des gonadotrophines d’ori-
gine hypothalamique. Les mutations de
TAC3 et TACR3 sont elles aussi respon-
sables d’HH congénital à odorat nor-
mal. Les mutations du gène LHb, indui-
sant un hypogonadisme sont très rares.
Par contre, plusieurs cas de mutation de
FSHb ont été décrits chez des femmes
avec aménorrhée primaire, estradiol bas
et une dissociation entre une FSH indé-
tectable et une LH élevée. Ces femmes
ont une adrénarche normale, mais une
absence de thélarche et de ménarche.
Leur fertilité peut être rétablie après ad-
ministration de FSH exogène.
LES HYPOGONADISMES
HYPOGONADOTROPES
SYNDROMIQUES SANS
ANOSMIE
Les hypogonadismes hypogonado-
tropes syndromiques sans anosmie sont
rares. Les mutations de DAX1 sont res-
ponsables d’une hypoplasie congénitale
des surrénales avec une insuffisance
surrénale néonatale, puis un déficit go-
nadotrope. Cette maladie est liée à l’X.
À ce jour, un seul cas de femme homo-
zygote DAX1, par duplication du gène,
avec une insuffisance gonadotrope, a
été rapporté. Cette patiente présentait
un HH isolé sans insuffisance surrénale.
Si l’hypogonadisme s’intègre dans un
tableau d’obésité, plusieurs étiologies
doivent être évoquées : le syndrome de
Prader-Willi, le syndrome de Bardet-
Biedl et les très rares mutations du gène
de la leptine et de son récepteur ou, de
manière encore plus exceptionnelle, des
mutations de la proconvertase de type 1.
LES HYPOGONADISMES
HYPOGONADOTROPES
FONCTIONNELS
Les hypogonadismes hypogonado-
tropes fonctionnels sont fréquents. Si
l’anorexie mentale est une cause clas-
sique d’hypogonadisme, les états de
dénutrition moins sévères, en particu-
lier les déficits de la masse grasse, sur-
tout dans le cadre d’activités sportives
intenses, sont souvent accompagnés de
déficit gonadotrope.
Les enfants atteints de maladie cœ-
liaque, de maladie de Crohn ou d’insuf-
fisance rénale chronique peuvent pré-
senter un impubérisme. L’utilisation
prolongée de glucocorticoïdes, tout
comme l’excès endogène (syndrome de
Cushing) est responsable d’un retard de
puberté par l’effet antigonadotrope du
cortisol sur la GnRH. Même si ce déficit
est réversible à l’arrêt de la corticothéra-
pie ou lors de la guérison du Cushing, il
doit être pris en compte comme facteur
potentiellement aggravant de l’ostéopo-
rose cortisonique.
CONCLUSION
Les causes génétiques d’hypogona-
dismes hypogonadotropes ont beau-
coup progressé depuis dix ans, même
si les mutations connues n’expliquent
encore que la moitié des HH.
L’utilisation des panels de gènes va
permettre l’amélioration du diagnos-
tic étiologique. L’identification des
gènes impliqués dans les HH permet
d’établir un conseil génétique et une
prise en charge précoce de la patho-
logie. Dans les formes partielles, diffi-
ciles à diagnostiquer, de GnRHR ou du
FGFR1, il faudra envisager systéma-
tiquement la possibilité que le déficit
gonadotrope soit réversible en faisant
une fenêtre thérapeutique. Les raisons
de cette réversibilité sont pour l’ins-
tant inconnues.
Cette observation appelle plusieurs
commentaires :
a) l’IRM hypophysaire est indispensable
devant un impubérisme à gonadotro-
phines basses,
b) un infléchissement statural évoquant
un retard pubertaire simple n’élimine
pas un hypogonadisme hypogonado-
trope congénital,
c) la classification des HH en fonction de
la présence ou non d’une anosmie n’est
pas parfaite !
✖L’auteur déclare ne pas avoir de liens d’intérêts.
MOTS-CLÉS
Hypogonadisme hypogonadotrope
congénital, Hypogonadisme hypogona-
dotrope fonctionnel, Hypogonadisme
hypogonadotrope isolé, Syndrome de
Kallmann
• Boehm U, Bouloux PM, Dattani MT
et al. European consensus statement
on congenital hypogonadotropic
hypogonadism : pathogenesis, diagnosis
and treatment. Nat Rev Endocrinol 2015;
11: 547-64.
• Bry-Gauillard H, Trabado S, Boulignad
J et al. Congenital hypogonadotropic
hypogonadism in females: clinical
spectrum, evaluation and genetics. Ann
Endocrinol 2010; 71: 158-62.
RÉFÉRENCES
1
/
3
100%