C`est une patho neurologique périphérique tronculaire qui

Pathologies neurologiques
BASE ANATOMO PHYSIOLOGIQUE EN NEURO
GENERALITE
L'organisation nerveuse, est construite sur un modèle de réseau, d'ou le nom de
système nerveux.
Cette organisation, peut se diviser en deux façons :
- Selon l'anatomie, système nerveux centrale, système nerveux périphérique.
- Selon la physiologie, système nerveux de relation et système nerveux végétatif.
Le système nerveux est composé de cellules dont certaines sont les cellules nobles ou neurone
et d'autres assurent le soutient la névroglie
LE NEURONE
Cette cellule a une structure particulière et est même macroscopique, certaine mesure
environ un mètre se long.
Cette cellule présente un corps cellulaire, des petits prolongements ou dendrites et
surtout un long prolongement ou axone.
Cet axone est protégé par des gaines de Myéline, de nombreuses pathologies graves ne
sont rien d'autre qu'une démyélinisation (exp-Sclérose en plaque.)
Cette cellule est extrêmement fragile devant tous les types d'agression :
- Manque d'oxygène
- Manque de glucose
- Tous les toxiques
De plus la souffrance neuronale sévère, entraîne une névrose, et globalement un
neurone détruit n'est pas remplacé.
Ceci explique la fréquence des séquelles neurologiques.
Pourtant il existe des récupérations neurologiques qui s'expliquent de plusieurs façon :
- Phénomène de remyélinisation
- Dans de nombreuses patho le déficit neuro est moins du à la destruction
neuronale qu'à l'œdème d'accompagnement, qui va régresser rapidement.
- En cas de section axonale précise, il peut y avoir réparation de l'axone. Cette
réparation peut être spontané ou par micro chirurgie ( suture axonale )
- Rééducation fonctionnelle, qui vise à mieux utiliser les réseau nerveux restant
ou les muscles nom paralysés.
Le neurone a un double fonctionnement, physique à type de fonctionnement électrique, grâce
à une dépolarisation.
Ce fonctionnement permet une exploration électrique
E.E.G = éléctro-encéphalo gramme
E.M.G = éléctro-myogramme
Vitesse de conclusion nerveuse.
Ceci permet également des traitements : éléctro-thérapie.
Un fonctionnement chimique, grâce à la fabrication de médiateurs chimiques (
Acéthylcholine, noradrénaline.)
Ces médiateurs permettent le passage de l'influx nerveux au niveau de la synapse par
traduction chimique.
Les neurones sont organisés en réseau grâce aux synapses. Mais il existe en plus des
inter-neurone qui ont souvent un effet inhibiteur, parfois à l'origine d'auto entraînement qui
serait à l'origine de l'installation des douleurs chroniques.

ORGANISATION DU SYSTEME NERVEUX :
SNC
C'est l'élément le plus important et donc le plus protégé.
Cette protection est double
- protection osseuse, avec le crâne et le rachis.
- protection membranaire, par les méninges ( Durmère, Arachoide, Pimère.)
Les méninges descendent dans le rachis, mais s'arrête bien avant la fin du rachis, mais
après la fin de la moelle épinière.
Ceci permet un geste, la ponction lombaire qui permet le recueil de liquide céphalo
rachidien sans danger pour les élément nerveux.
Le S.N.C. comprend les éléments suivants :
- Le cerveau
- Le cervelet
- Le tronc cérébral
- La moelle épinière
LE CERVEAU
Il est constitué de deux hémisphère cérébraux reliés entre eux, d'ou une commande
séparée de chaque hémi-corps.
Et d'ou la fréquence des hémiplégie.
Pour la plupart des informations, cette commande est croisée, car il existe un
phénomène de décussation surtout dans le tronc cérébral.
Chaque hémisphère est de plus spécialisé :
- L'hémisphère gauche contrôle le langage c'est à dire la parole, l'écriture et la lecture.
- L'hémisphère droit, est spécialisé dans la reconnaissance de l'image corporelle.
Les hémisphères cérébraux présentent du liquide céphalo-rachidien, d'ou des
possibilités d'exploration et des pathologies d'accumulation du L.C.R, c'est l'hydrocéphalie.
Les différentes régions du corps sont représenté au niveau du cerveau ainsi que les
types d'activité neurologique. (Sensibilité motricité "fronto-latéral").
Cette zone capitale représente la surface du cerveau ou le cortex cérébral.
Dans la profondeur des hémisphères, il existe des zones particulières identifiées, comme les
noyaux gris du cerveau (Thalanus…)
Il existe enfin, une zone indépendante du cerveau, mais en connexion avec ce dernier c'est
l'hypophyse (glande intracranienne.)
LE CERVELET
Il est composé de deux hémisphères cérébelleux reliés par un vermis cérébelleux, il est
situé dans un logement en arrière du crâne ( la fosse postérieur)
Ce cervelet est irrigué par le tronc vertébral basilaire qui est souvent victime de
pathologie à type d'insuffisance vertébral-basilaire, source de malaise, surtout chez le sujet
âgé. Le cervelet joue deux rôles essentiels :
- Contrôle de l'équilibre, en association avec l'oreille interne, la vision et les récepteurs
articulaires.
- Contrôle de l'exécution du mouvement, permettent l'assemblage de gestes simples, en un
mouvement complexe.

Quand il existe, une atteinte du cervelet, il apparaît un syndrome cérébelleux avec
surtout, trouble de l'équilibre et trouble de la coordination des mouvements dans l'espace et
dans le temps.
LE TRONC CEREBRAL
Cette structure correspond à la convergence des éléments nerveux centraux pour
former une structure cylindrique à la base du crâne, précèdent le cordon médullaire, qui
descendra dans le rachis.
Cette structure est donc très fragile et ces pathologies sont particulièrement sévères.
Certaines voies neurologiques vont croisées au niveau du tronc cérébral, c'est la
décussation.
Une atteinte du tronc cérébral peut donc entraîné des symptômes sur chaque hémi-corps, se
sont les syndromes alternés.
Au niveau du tronc cérébral, sont situés des centres neurologiques dont certains
contrôles des fonctions vitales, c'est le cas du bulbe rachidien qui contrôle en particulier la
ventilation.
Ceci explique les arrêts ventilatoires du coup du lapin, la mort subite du nouveau née
et les intoxications médicamenteuses et les accidents de toxicomanie.
Ce tronc cérébral, donne naissance à des nerfs crâniens ( 12 paires) qui contrôlent les
fonctions sensorielles, la motricité faciale, la sensibilité facile, et l'occulo-motricité.
LA MOELLE EPINIERE
La moelle épinière est à la fois un élément du syst nerv central, et du système nerv
périphérique.
Sa partie S.N.C. est au niveau de la substance blanche donc à la périphérie de la
moelle épinière.
Les éléments médulaires S.N.P. sont situés dans la partie central de la moelle sous la
forme de la substance grise avec des cornes antérieurs et postérieurs.
La moelle dans son rôle centrale n'est rien d'autre qu'un système relais entre le cerveau
et le S.N.P.
Elle transmet de façon descendante la motricité et de façon ascendante sensibilité.
La moelle dans son rôle périphérique ( substance grise) est le lieu de relais entre le
premier neurone et le deuxième neurone. C'est le lieu des synapses surtout motrice.
Cette moelle ( grise) contrôle la motricité involontaire c'est à dire le tonus ( état de contraction
permanente au repos) et les réflexes.
Quand il survient une section médullaire, les 2 parties de la moelle sont atteintes.
L'interruption des voies centrales ( blanche) va entraîner un déficit de motricité volontaire,
bilatéral en dessous de l'étage de la lésion (paraplégie, section au niveau du dos) en même
temps s'installe une anesthésie bilatéral au dessus de la lésion.
De plus s'ajoute des anomalies de la motricité involontaire, cette motricité involontaire
s'exagère car la moelle devient autonome libérée de l'inhibition cérébral, avec augmentation
du tonus, augmentation des réflexes et même syncinésies c'est à dire des mouvement parasite
involontaire déclenché par un mouvement volontaire dans le territoire non paralysé.
La moelle épinière est protégée par des méninges avec présence de liquide céphalo-
rachidien toute pathologie médullaire suspectée impose donc un geste (la ponction lombaire
permettant l'étude du L.C.R.)
De la moelle épinière naissent des racines nerveuses à chaque étage vertébrale de façon
bilatérale et ces racines se continuent par des troncs nerveux. Se sont les nerfs rachidiens.

Ces nerfs rachidiens naissent de la substance grise de la moelle. Les racines
antérieures naissent dans les cornes antérieurs de la moelle et sont motrices. Les racines
postérieurs naissent dans les cornes postérieurs et transmettent la sensibilité. ( S.N.P.)
Chaq racine ant rejoint une racine post et la fusion des deux donnent un tronc nerveux.
S.N.P.
Il comprend les racines nerveuses, les troncs nerveux et les terminaisons nerveuses,
mais également la substance grise de la moelle épinière.
Les racines nerveuses contrôle tout un territoire du corps sur le plan moteur et sensitif.
Ces territoires sont appelés métameres et au niveau du tronc, il s'agit d'étages en
ceinture ou plutôt en hemi-ceinture, ceci explique les névralgies inter-costale au niveau des
membres, chaque racines contrôle un trajet qui va du début du membre jusqu'a l'extrémité
(Sciatique douleur de la fesse jusqu'au pied).
Les troncs nerveux s'acheminent dans tout l'organisme et présentent des terminaisons
dont certaines sont motrices au niveau du muscles sous le forme de plaque motrice, d'autres
terminaisons sont sensitives et ces terminaisons sont différentes pour chacune des sensibilités.
- Profonde (fuseau neuro-musculaire)
- Superficielle :
- algésique (en surface)
- thermique
- superficielle tactile
Ceci explique la possibilité de trouble sensitif dissocés.
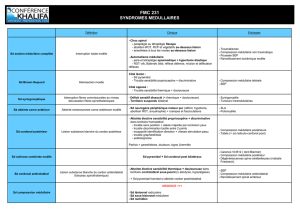
LES GRANDS SYNDROMES NEUROLOGIQUES :
Ils sont nombreux et certains sont centraux et d'autres périphérique.
Pour les syndromes centraux c'est :
- Le syndrome pyramidale.
- Le syndrome extra pyramidale (PARKINSON)
- Le syndrome cérébelleux
- Le syndrome d'hypertension intra-cranienne
- Le syndrome méningé
Pour les syndromes périphériques c'est :
- Le syndrome neurogène périphérique
- Le syndrome radiculaire ( SCIATIQUE)
- Le syndrome tronculaire (POLYNEVRITE)
- Le syndrome canalaire (CANAL TARSIEN, MORTONE)
SYNDROMES CENTRAUX
SYDROME PYRAMIDALE
Sa découverte affirme une atteinte centrale.
Comme les voies pyramidales ne conduisent que la motricité, ce syndrome est
uniquement moteur.
Les symptômes sont les suivants :
*Déficit de motricité volontaire, plus ou moins intense, soit déficit partiel moteur ou
parésie soit déficit totale ou paralysie vraie (une plégie.)

*Exagération du tonus ou hypertonie qui est particulière, c'est l'hypertonie élastique
responsable de spasticité, avec résistance augmentant lors d'un mouvement passif et retour à
la position initiale quand on fait cesser le mouvement.
*Exagération des réflexes ostéo-tendineux ou hyperéflectivité, avec réflexe vif, ample,
diffusé (dix centimètre au dessus ou en dessous de la zone) polycinétique (plusieurs
mouvements après la stimulation)
*Anomalie des réflexe cutanés (R.C.P).
Le R.C. plantaire qui se fait normalement et en flexion plantaire des orteils, va se faire en
extension (F.D.) des orteils plus écartement des orteils, ce R.C.P inversé est le signe de
BABINSKI.
Ce signe de BABINSKI peut être le seul signe du syndrome pyramidale à son début.
*En plus apparition de sincinésies mouvement involontaire dans le territoire paralysé.
SYNDROME EXTRA PYRAMIDALE ( PARKINSON)
Les voies extra pyramidale font partit du S.N.C. et sont organisés sous la forme d'un
réseau avec des centres spécifiques (les noyaux gris centraux).
Les principaux noyaux sont :
Le thalamus
Locus niger
Le striatum
Le pallidum
Une pathologie répandue, correspond à l'atteinte de deux noyaux gris le locus niger et
le striatum, cette atteinte nigrostriée est responsable de la maladie de Parkinson.
Ce syndrome extra pyramidale est souvent appelé le syndrome parkinsonien.
Les symptômes sont les suivants :
*Augmentation du tonus ou hypertonie, mais cette hypertonie est plastique, avec résistance
pendant un mouvement passif à type de tuyau de plomb et avec phénomène de roue dentée et
à la fin du mouvement passif le segment mobilisé reste dans la position terminale.
*Difficulté pour réaliser les mouvements avec lenteur des mouvements, retard à l'initiation,
caractères saccadés des mouvements.
Ces phénomènes portent sur la marche, "marche à petit pas lent et sacadé", les mouvements
des membres supérieurs, la parole "élocution lente et scandée", l'écriture "micrographie" et sur
la mimic "visage inexpressif, fasciés figée"
*Tremblement, particulier maximum au repos, diminué à l'effort, majoré par l'émotion et
avec gestes complexes à type d'émiettement.
*Les réflexes ostéo tendineux peuvent être normaux ou vif.
*Troubles trophiques avec rétraction des tissus capsulo-ligamentaire surtout à la main
mais également aux pied.
Dans la plupart des cas le syndrome extra pyramidale peut être assimilé à un syndrome
parkinsonien.
La plupart des syndromes parkinsonien sont primitif survenant chez le sujet âgé, c'est
alors la maladie de parkinson.
Dans certain cas le syndrome parkinsonien est secondaire ( intoxication au Co oxyde
de carbone) infection cérébrale, rarement d'A.V.C.
SYNDROME CEREBELEUX
Les signent sont les suivants :
*Difficulté de coordination des mouvements dans l'espace et dans le temps.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
1
/
55
100%