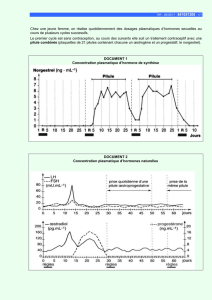
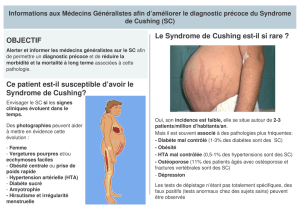
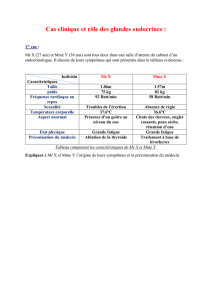
Télécharger le texte intégral

ÉCOLE NATIONALE VÉTÉRINAIRE D’ALFORT
Année 2013
SÉCRÉTIONS EN EXCÈS D’HORMONES
SEXUELLES ET DE PRÉCURSEURS
STÉROÏDES PAR LES CORTICOSURRÉNALES.
Vers de nouveaux outils biologiques, vers de nouvelles formes
clinico-biologiques d’hypercorticisme chez le chien et chez le chat ?
Revue bibliographique
THÈSE
Pour le
DOCTORAT VÉTÉRINAIRE
Présentée et soutenue publiquement devant
LA FACULTÉ DE MÉDECINE DE CRÉTEIL
le……………
par
Stéphanie TOROK
Née le 5 août 1978 à Paris 16ème
JURY
Président : Pr.
Professeur à la Faculté de Médecine de CRÉTEIL
Membres
Directeur : M. Dan Rosenberg
Maître de conférences à l’Ecole Nationale Vétérinaire d’Alfort
Assesseur : Mme Hélène Combrisson
Professeur à l’Ecole Nationale Vétérinaire d’Alfort


REMERCIEMENTS
Au Professeur de la faculté de Médecine de Créteil,
Pour nous avoir fait l’honneur d’accepter la présidence de notre jury de thèse.
Hommage respectueux.
A Monsieur Rosenberg,
Maître de Conférences à l’Ecole Nationale Vétérinaire d’Alfort,
Pour avoir accepté d’encadrer et de corriger ce travail et de participer à notre jury de thèse.
Sincères remerciements.
A Madame Combrisson,
Professeur à l’Ecole Nationale Vétérinaire d’Alfort,
Pour avoir accepté de corriger ce travail et de participer à notre jury de thèse.
Sincères remerciements.

A mes parents et à ma sœur. Je vous aime.
A mon arrière-grand-mère qui me manque.

1
TABLE DES MATIERES
Table des illustrations........................................................................................................7
Tableaux....................................................................................................................................7
Figures.......................................................................................................................................9
Liste des abréviations.......................................................................................................11
Avant-propos......................................................................................................................13
INTRODUCTION.............................................................................................................15
1. Rappels d’anatomie et de physiologie des corticosurrénales......................17
1.1. Anatomie, embryologie et histologie fonctionnelle des
corticosurrénales.....................................................................................................17
1.2. Physiologie des corticosurrénales ......................................................................19
1.2.1. Stéroïdogenèse corticosurrénalienne.......................................................................19
1.2.1.1. Équipement cellulaire spécifique de la corticosurrénale..........................................19
1.2.1.2. Les différentes voies de la stéroïdogenèse corticosurrénalienne..............................20
1.2.2. Régulation de sécrétion des hormones stéroïdes par les corticosurrénales .........24
1.2.2.1. Régulation de sécrétion des glucocorticoïdes et des androgènes surrénaliens.........24
1.2.2.2. Régulation de sécrétion des minéralocorticoïdes.....................................................26
1.2.3. Principaux effets biologiques des hormones stéroïdes...........................................28
1.2.3.1. Mode d’action des hormones stéroïdes - récepteurs stéroïdiens..............................28
1.2.3.2. Effets biologiques des glucocorticoïdes...................................................................30
1.2.3.3. Effets biologiques des minéralocorticoïdes..............................................................31
1.2.3.4. Effets biologiques des androgènes surrénaliens.......................................................32
1.2.4. Transport sanguin des hormones stéroïdes ............................................................32
1.2.5. Catabolisme des hormones stéroïdes.......................................................................34
2. Facteurs à prendre en compte pour l’interprétation et la justification
de prescription d’un test hormonal.....................................................................35
2.1. Performances techniques d’un test hormonal ..............................................35
2.1.1. Méthodes de dosage des hormones stéroïdes..........................................................35
2.1.2. Caractéristiques analytiques des dosages d’hormones stéroïdes..........................37
2.2. Intervalles de référence.........................................................................................39
2.3. Qualités intrinsèques d’un test diagnostique : sensibilité, spécificité...40
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
1
/
206
100%