4 n° Re p è re s

Fiche à détacher
diapositives
téléchargeables
La Lettre du Cancérologue • Vol. XX - n° 10 - décembre 2011 | I
Repères
biologiques
Cible • Signal • Patient 1
n° 4
1 Fiche sous la responsabilité de ses auteurs.
Nouvelle cible, nouvelle entité clinico-
biologique et nouvelle thérapeutique :
l’exemple d’ALK dans les cancers
bronchiques non à petites cellules
C. Massard*, B. Besse*, D. Planchard*
* Département de médecine oncologique, service d’innovation thérapeutique et essais
précoces (SITEP), institut de cancérologie Gustave-Roussy, Villejuif.
L
a découverte de certaines anomalies moléculaires peut
radicalement modifier la stratégie thérapeutique d’un
cancer ; elle peut aussi plus profondément modifier la stratégie
de prise en charge et de diagnostic des patients ayant un
cancer. Ainsi, les anomalies de KIT ont permis de faire émerger
la pathologie des tumeurs stromales gastro-intestinales
(
GastroIntestinal Stromal Tumor
[GIST]) comme une entité à
part entière, et cette pathologie orpheline des années1990
est devenue le symbole et le paradigme de la cancérologie du
xxiesiècle. Récemment, les progrès dans la compréhension de
la biologie moléculaire des cancers bronchiques, en particulier
de ceux non à petites cellules (CBNPC), ont permis d’identifier
différentes altérations moléculaires, qui sont autant de cibles
thérapeutiques potentielles, mais aussi potentiellement des
outils pour mieux diagnostiquer les CBNPC, et permettre d’en
changer la vision. Cette approche à la carte nous entraîne de
plus en plus vers la possibilité d’individualiser le traitement
selon le profil pathologique etmoléculaire des patients. En
effet, il n’existe probablement pas 1entité homogène de
CBNPC, mais plutôt une addition de maladies “rares”, avec des
anomalies moléculaires particulières, des voies de signalisation
activées distinctes, une évolution clinique singulière et des
potentialités thérapeutiques différentes. Plusieurs tests molécu-
laires génomiques validés, réalisés à partir du tissu tumoral,
font maintenant partie de la prise de décision thérapeutique
pour le cancer du poumon. Cette individualisation moléculaire
est récente et fait suite à l’individualisation de populations
bénéficiant des inhibiteurs de tyrosine kinase (ITK) de l’EGFR
(Epidermal Growth Factor Receptor)
, la première étant celle des
sujets non fumeurs présentant un profil oncogénique particulier.
Ce cancer apparaît donc comme une maladie du génome.
Ainsi, la détection des mutations EGFR
,
KRAS
,
HER2
(Human
Epidermal growth factor Receptor-2)
ou des réarrangements
comme ALK
(Anaplastic Lymphoma Kinase)
vont permettre
dans les années à venir d’avoir une nouvelle classification des
CBNPC : il ne sera bientôt plus possible de parler de “cancer
du poumon”, en général, comme il n’est plus possible de
parler de sarcome ou de cancer du sein ; il sera indispen-
sable de déterminer ces anomalies moléculaires pour savoir de
quelle maladie souffre un patient donné, et pour décider de la
meilleure stratégie thérapeutique
(tableauxI etII, p.II)
[1]
.
LK12-fiche.indd 1 09/12/11 15:18

Fiche à détacher
II | La Lettre du Cancérologue • Vol. XX - n° 10 - décembre 2011
Repères biologiques n° 4
Une nouvelle cible :
ALK dans les cancers du poumon...
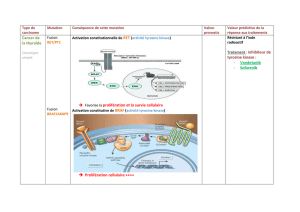
La protéine ALK est un récepteur à activité tyrosine kinase
initialement identifié dans les lymphomes anaplasiques dans
les années1990. En2007, une équipe japonaise a identifié
pour la première fois une fusion des gènes ALK avec EML4
(Echinoderm Microtubule-associated protein-Like4)
dans
les CBNPC. Cette anomalie est due à une translocation au
niveau du chromosome2p
(2)
. Différents variants sont décrits.
La protéine de fusion “chimétique” EML4-ALK possède des
propriétés oncogéniques dans des modèles in vivo et in vitro
,
avec une activation constitutionnelle de la voie de signalisation
ALK passant par la voie des MAPK
(Mitogen-Activated Protein
Kinase)
et PI3K (phosphoinositide 3-kinase)/AKT, aboutissant à
un bénéfice en termes de survie et de prolifération. L’inhibition
de l’activité kinase ALK par des inhibiteurs pharmacologiques
entraîne l’apoptose des cellules tumorales EML4-ALK positives,
et une diminution des tumeurs dans des modèles murins.
Ces réarrangements génomiques du gène ALK sont détectés
chez3à7 % des patients porteurs d’un CBNPC. Comme pour les
mutations EGFR, les réarrangements ALK sont plus fréquemment
retrouvés chez les patients ayant un adéno carcinome et chez les
sujets non ou peu fumeurs. De plus, il semble, dans les premières
études, que les réarrangements ALK soient un phénomène spéci-
fique des mutations EGFR ou KRAS.
... mais aussi dans d’autres pathologies
Les réarrangements ALK sont aussi présents dans une pathologie
tumorale mésenchymateuse rare, les tumeurs myofibroblastiques
inflammatoires, qui concernent le plus souvent des patients
jeunes. Ces tumeurs sont essentiellement de présentation pulmo-
naire ou ganglionnaire. La biopsie montre des cellules myofibro-
blastiques avec une réaction stromale importante. Le traitement
de choix est chirurgical, sachant que ces tumeurs possèdent un
faible potentiel métastatique. Dans plus de la moitié des cas,
il est montré des altérations d’ALK et des réponses tumorales
sont observées avec le crizotinib.
Un nouveau traitement
(figure1)
Étant donné que l’activité kinase est importante dans le
processus de carcinogenèse, il était logique de tester des ITK
ciblant la voie de signalisation ALK. Cependant, le crizotinib
a été initialement développé comme un inhibiteur de MET.
Ila démontré, dès la phaseI, une activité clinique majeure et
très impressionnante chez des patients porteurs d’un CBNPC
qui présentaient un remaniement ALK. Après avoir étudié plus
de 1 500patients porteurs d’un CBNPC, E.L.Kwak etal. ont
inclus 82patients transloqués ALK dans cette étude d’extension
de phaseI, testant le crizotinib en monothérapie chez des
sujets déjà polytraités (41 % avec au moins 3lignes de traite-
ments). Les résultats sont impressionnants, avec un taux de
réponse partielle de 57 % (47patients sur82 avaient une
réponse partielle et une réponse complète), un taux de survie
sans progression à 6mois de 72 % et une durée moyenne
du traitement de 6,4mois (données encore non matures).
Le crizotinib a peu d’effets secondaires (principalement des
toxicités digestives et des troubles visuels). Cette étude a été
une étape majeure dans le développement de ce médicament,
mais aussi en général en oncologie thoracique
(3)
. Il est donc
apparu très vite que cette activité antitumorale était restreinte
aux patients ayant un CBNPC avec un réarrangement ALK,
et qu’il était donc indispensable de mettre en place un
diagnostic moléculaire enparallèle de cette étude pour
Tableau II. Altérations moléculaires, cancer du poumon
et opportunité thérapeutique.
Molécules Fréquence (%)
Mutation de l’EGFR Erlotinib, géfitinib
Nouveaux pan-HER
10
Translocation d’EML4-ALK Crizotinib 3 à 5
Altération de HER2 Trastuzumab (mutation),
PF-299804 (amplification)
2
2
Mutation de PI3K GDC-0941, XL147
XL765, PX-866
BEZ235, BKM120
2 à 33
Amplification/mutation
deMET
XL184, ARQ 197, MetMAb 5
Mutation de RAS et RAF Sorafénib, GSK1120212
AS703026, AZD6244
3 à 30
Amplification de FGFR1 BJG398, AZD4547, TKI258 10 à 20
Tableau I. Altérations moléculaires et cancer du poumon.
Épidermoïde Adénocarcinome
Mutation de KRAS (%) 5 10 à 30
Mutation de BRAF (%) 3 2
EGFR (%)
Mutation de tyrosine kinase
Amplification
Mutation variant III
2 à 5
30
5
10 à 40
15
Rare
HER2 (%)
Mutation de tyrosine kinase
Amplification
Rare
2
2 à 4
6
Fusion d’ALK (%) Rare 4 à 7
MET (%)
Mutation
Amplification
5
< 10
5
< 10
Amplification de FGFR1 (%) 20 1
Mutation de LKB1 (%) 10 à 20 30 à 40
PIK3CA (%)
Mutation
Amplification
2
33
2
6
LK12-fiche.indd 2 09/12/11 15:18

Fiche à détacher
Figure 1. Taux de réponse des patients avec un cancer bronchique non à petites cellules traité par crizotinib.
60
Diminution de la taille tumorale (%)
20
40
0
– 20 – 30 %
OR = 57 %
Maladie progressive
Maladie stable
Réponse partielle confirmée
Réponse complète confirmée
– 40
– 60
– 80
– 100
La Lettre du Cancérologue • Vol. XX - n° 10 - décembre 2011 | III
Repères biologiques n° 4
identifier les patients présentant l’altération moléculaire
(3,4)
.
La technique de référence pour la détection d’un remaniement
ALK est actuellement la technique par hybridation in situ en
fluorescence (FISH), sachant que d’autres méthodes existent et
sont en cours de validation dans le CBNPC, telles que l’immuno-
histochimie ou le séquençage direct du gène après amplifi-
cation. Deux grandes phasesIII d’enregistrement comparant
le crizotinib à une chimiothérapie en première et en seconde
ligne de traitement chez les patients ALK-positifs sont en cours.
Compte tenu de l’activité antitumorale sans précédent du
crizotinib chez ces patients, la FDA (Food and Drug Adminis-
tration) a décidé récemment de ne pas attendre les résultats
des études de phaseIII (qui se poursuivent en Europe) et
d’approuver sa commercialisation, faisant du crizotinib l’un
des médicaments anticancéreux dont le développement a été
le plus rapide. Il est à noter, par ailleurs, que la présence d’un
remaniement ALK est plutôt un facteur de mauvais pronostic
comparativement aux patients sans anomalie ALK et à ceux
porteurs d’une mutation EGFR
(4).
L’avenir
Compte tenu de l’activité du crizotinib, les oncologues thoraciques
auront à faire face à différents défis dans les années à venir.
Premièrement, il est aujourd’hui devenu nécessaire de pouvoir
proposer la recherche de l’anomalie ALK chez des patients ayant
un CBNPC. La technique de référence dans le CBNPC est actuel-
lement la technique par FISH, mais d’autres techniques plus
“exportables” devraient se mettre en place, comme notamment
l’immunohistochimie. Le cancer bronchique, comme beaucoup
d’autres cancers, est entré dans l’ère de la thérapie à la carte.
Le blocage de la voie ALK chez les patients présentant une
activation conduit à un bénéfice clinique majeur. Il paraît indis-
pensable à ce jour que les patients porteurs d’un CBNPC puissent
avoir accès à un diagnostic moléculaire comportant au minimum
une recherche des mutations EGFR et des réarrangements ALK,
compte tenu du fait que ces pathologies sont clairement des
entités clinicobiologiques différentes, avec des stratégies théra-
peutiques elles aussi radicalement différentes. Il sera cependant
important de mettre en place des recommandations nationales
et internationales pour le suivi et la stratégie diagnostique du
CBNPC dans les années à venir.
Deuxièmement, les patients qui répondent au crizotinib vont
malheureusement développer des résistances secondaires à
cette molécule, qui commencent à être en partie comprises. Ces
mécanismes de résistance font intervenir des mutations secon-
daires de résistance ou des activations de voies de signalisation
parallèles telles que la voie de l’EGFR. Un des défis majeurs sera
de définir précisément quels mécanismes de résistance sont
en jeu chez un patient donné, et de développer de nouvelles
stratégies pharmacologiques avec des agents de nouvelles
générations ou avec des combinaisons thérapeutiques (par
exemple, crizotinib et inhibiteur de l’EGFR)
[5-7]
.
Enfin, comme la plupart des thérapies ciblées, le crizotinib
ne permet pas de guérir les patients porteurs d’un CBNPC à
un stade avancé. Il sera sans doute intéressant de tester ce
médicament dans les stades moins avancés, en particulier
en adjuvant.
LK12-fiche.indd 3 09/12/11 15:18

Fiche à détacher
Figure 2. Stratégie thérapeutique en première ligne pour un patient avec un cancer bronchique non à petites cellules.
Non SCC
Histologie
Non Oui
Addiction oncogénique
SCC
EGFR : géfitinib/erlotinib
ou
ALK : crizotinib
Doublet de platine +
bévacizumab
ou
platine + pémétrexed
Doublet de platine
Maintenance : oui/non
IV | La Lettre du Cancérologue • Vol. XX - n° 10 - décembre 2011
Repères biologiques n° 4
Conclusion
(figure 2)
La découverte des réarrangements ALK en 2007 a permis
l’identification d’un sous-type moléculaire de CBNPC. Les
premiers essais ont démontré dès2009 l’intérêt de cibler cette
anomalie avec des ITK comme le crizotinib, qui a été approuvé
cette année par la FDA. L’histoire du crizotinib est l’une des plus
rapides, elle représente l’une des plus spectaculaires réussites
dans le développement clinique d’une stratégie anticancéreuse
et devient un exemple de la médecine personnalisée. ■
1. Sasaki T, Jänne PA. New strategies for treatment of ALK
rearranged non-small-cell lung cancers. Clin Cancer Res
2011. [Epub ahead of print]
2. Soda M, Choi YL, Enomoto M et al. Identification of
the transforming EML4-ALK fusion gene in non-small-cell
lung cancer. Nature 2007;448(7153):561-6.
3. Kwak EL, Bang YJ, Camidge DR et al. Anaplastic
lymphoma kinase inhibition in non-small-cell lung cancer.
N Engl J Med 2010;363(18):1693-703.
4. Shaw AT, Yeap BY, Mino-Kenudson M et al. Clinical
features and outcome of patients with non-small-cell lung
cancer who harbor EML4-ALK. J Clin Oncol 2009;27(26):
4247-53.
5. Choi YL, Soda M, Yamashita Y et al; ALK Lung Cancer
Study Group. EML4-ALK mutations in lung cancer that
confer resistance to ALK inhibitors. N Engl J Med 2010;
363(18):1734-9.
6. Sasaki T, Koivunen J, Ogino A et al. A novel ALK
secondary mutation and EGFR signaling cause resis-
tance to ALK kinase inhibitors. Cancer Res 2011;71(18):
6051-60.
7. Shaw AT, Yeap BY, Solomon BJ et al. Effect of crizotinib
on overall survival in patients with advanced non-small-
cell lung cancer harbouring ALK gene rearrangement: a
retrospective analysis. Lancet Oncol 2011;12(11):1004-12.
Références bibliographiques
Téléchargez les figures de cette fiche sur notre site internet
www.edimark.fr
> La Lettre du Cancérologue > Sommaire de décembre 2011
LK12-fiche.indd 4 09/12/11 15:18
1
/
4
100%