resume chimie orga eduscol

Stéréochimie
1- Des stéréoisomères (ou isomères de configuration) sont des composés de
même formule brute, mais qui diffèrent seulement par la disposition des
atomes dans l'espace. Les stéréoisomères correspondent à des
configurations différentes.
2- Une molécule de configuration donnée est chirale si elle est non
superposable à son image dans un miroir plan (elle est dépourvue de plan et
de centre de symétrie). Deux énantiomères (ou inverses optiques ou
antipodes optiques) sont donc symétriques l'un de l'autre dans un miroir ET
non superposables.
3- Un carbone asymétrique, noté C*, est un carbone tétraédrique (sp3), dont
les 4 substituants sont tous différents.
4- L'existence d'UN ET d'UN SEUL carbone asymétrique est une condition
suffisante de chiralité. Elle n'est pas du tout nécessaire: Voir 5-
5- Il existe des molécules sans carbone asymétrique qui sont chirales : certains
allènes, certains spiranes, les biphényls orthodisubstitués (atropoisomérie)
… par exemple.
Il existe des molécules qui possèdent plusieurs carbones asymétriques et qui
ne sont pas chirales : composés méso par exemple. Voir 14-
6- La configuration relative d'un carbone asymétrique est déterminée par les
règles de Cahn-Ingold-Prelog (C.I.P.) qui établissent un ordre de priorité
conventionnel entre les 4 substituants différents du C*.
7- Une molécule est optiquement active (ou possède un pouvoir rotatoire)
si elle fait dévier le plan de polarisation de la lumière polarisée ; vers la
droite la molécule est dite dextrogyre, noté (d) ou (+) ; vers la gauche elle
est dite lévogyre, (l) ou (-)
8- Principe de Pasteur : une molécule est optiquement active si et seulement si
elle est chirale.
9- Il n'y a AUCUNE relation entre la configuration relative et le signe du
pouvoir rotatoire : un composé (R) peut être lévogyre ou dextrogyre ; mais
si le composé chiral est dextrogyre, son énantiomère est nécessairement
lévogyre.
De même la nomenclature (D) et (L) n'a PAS de rapport avec le signe du
pouvoir rotatoire. De plus il n'y AUCUN rapport entre (R)/(S) et (D)/(L).
10- Un mélange équimolaire de 2 énantiomères est optiquement inactif (par
compensation) : c'est un mélange racémique. Symbole :()±
11- Deux énantiomères : - ont les mêmes distances interatomiques et donc les
mêmes constantes physiques : température de
changement d'état, densité …
- n'ont pas la même action sur la lumière polarisée :
leurs pouvoirs rotatoires sont identiques en valeur
absolue, mais opposés.
- ont les mêmes propriétés chimiques vis à vis d'un
réactif achiral.
- ont des comportements chimiques différents vis à
vis d'un réactif chiral (une chaussure droite
distingue un pied droit d'un pied gauche, alors
qu'une chaussette ne le fait pas !). Ceci induit des
comportements biologiques très différents.
12- Des diastéréoisomères sont des stéréoisomères qui ne sont pas
énantiomères. Cela n'a donc rien à voir avec l'isomérie optique : un
diastéréoisomère donné peut être chiral ou achiral.
13- Un premier exemple de diastéréoisomérie (sans qu'il y ait isomérie
optique) est la stéréoisomérie de type Z, E due à une double liaison
carbone-carbone. On parle de stéréoisomérie géométrique.
14- Un second exemple de diastéréoisomérie est due à la présence de 2
carbones asymétriques dans une molécule. Si les 2 C* sont différemment
substitués, il y a 4 stéréoisomères tous chiraux formant 2 couples
d'énantiomères (le couple érythro et le couple thréo).
Attention : dans le cas où les 2 C* sont identiquement substitués, il n'y
a que 3 stéréoisomères, qui sont le couple thréo (les deux sont chiraux),
et le composé méso (achiral : existence d'un plan de symétrie).
Chaque énantiomère érythro (ou le méso) est diastéréoisomère de chaque
énantiomère thréo.
Dans le cas des cyclanes disubstitués, on a la nomenclature cis-trans.
15- Des diastéréoisomères : - n'ont pas les mêmes propriétés chimiques
- n'ont pas les mêmes propriétés physiques ; cette
différence permet de dédoubler le racémique par
réaction avec un composé chiral (formation de
deux diastéréoisomères que l'on pourra donc
séparer).

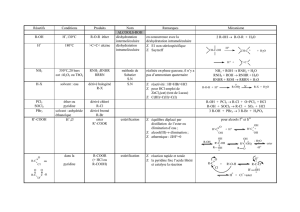
DERIVES HALOGENES ALIPHATIQUES R-X
Réactifs Conditions Produits Nom Remarques Mécanisme
R’O-Na+solvant : alcool ou
DMSO R-O-R’
éther synthèse de
Williamson éliminations sur R fréquentes R-I + R’-ONa → R-O-R’ + NaI
NH3
R-NH2
R1-NH-R2
R1R2R3N
solvant : eau ou
éther amines et sel
d’ammonium
quaternaire
synthèse
d’Hofmann • obtention de mélanges
d’amines et d’ammonium
quaternaire
• séparation par distillation
• on peut ajouter une base
(HCO3- ou CO32-) pour
déprotoner les ions
ammonium et générer les
amines
NH3 + RX R-NH3 X R-NH2 + NH4 X
R-NH2 + RX NH2 X NH + R-NH3 X
+- +-
+-+-
RR
RR
NH3
R-NH2
NH + RX NH X N-R + NH X
R
RR
R
R
+ - R
R
R
R
+ -
N-H
R
R
R-N + RX R-N-R X
R
R
R
R
+-
KCN solvant :
acétone ou DMF R-C≡N
nitrile S.N l’hydrolyse acide fournit l’acide
R-COOH
passage R-X→R-COOH
R-I + C≡N- → R-C≡N + I-
R-C≡N + H3O+ + H2O → R-COOH + NH4+
CH2(COOEt)2EtONa puis
H3O+ et ∆R-CH2-COOH synthèse
malonique
S.N
passage R-X→R-CH2-COOH H2C
COOEt
COOEt + B HC COOEt
COOEt + BH
--
HC
COOEt
COOEt
-+ R-X + X-
R-CH
COOEt
COOEt
R-CH
COOEt
COOEt
OH-R-CH
COO-
COO-R-CH2-COOH + CO2
H+
∆
Li pentane,-10°C R-Li réaction de Ziegler formation d’un
organolithien R-X + 2 Li → R-Li + LiX
Mg éther, 35°C R-MgX réaction de
Grignard formation d’un organomagnésien R-X + Mg → R-MgX
Na hexane, 60°C R-R duplication
de Wurtz • passage par l’organosodique
• utilisation de R-I R-I + 2 Na → R-Na + NaI
R-Na + R-I → R-R + NaI
Zn CH3COOH R-H réduction possible aussi avec LiAlH4,éther 2 Zn + 2 H+ + 2 RX → Zn2+ + ZnX2 + 2 R-H

DERIVES HALOGENES AROMATIQUES Ar-X
Réactifs Conditions Produits Nom Remarques Mécanisme
Nu-∆substitution de
X par Nu S.N • mécanisme addition-
élimination
• nécessité de groupements
attracteurs en o et p
N
N
X
O2
O2
+ Nu-N
N
XNu
-
stabilisé par mésomérie par NO2
X +
-N
N
Nu
O2
O2
O2
O2
NH2-NH3 liq substitution de X
par NH2
S.N • mécanisme élimination-
addition
• intermédiaire benzyne
• les autres groupes présents
orientent à l’inverse des
règles de Hollemann
X
*+ NH2
-X + NH3 + *
benzyne
-
NH3
++
NH3
+
NH3**
NH2
*
NH2
*
Mg solvant : éther Ar-MgX Ar-Br + Mg → Ar-MgBr
Na + R-I solvant :hexane Ar-R réaction de
Wurtz-Fittig utilisation de R-I Ar-I + R-I + 2 Na → 2 NaI + Ar-R
Cu ∆Ar-Ar réaction de
Ullmann utilisation de Ar-I 2 Ar-I + 2 Cu → Ar-Ar + 2 CuI

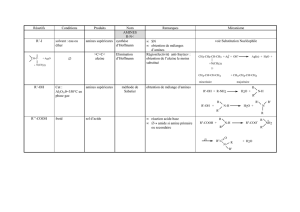
Préparations des dérivés halogénés
1- ALIPHATIQUES
Substrat Réactifs Conditions Produits Nom Remarques Mécanisme
R-H Cl2, Br2hν ou ∆R-H + H-X halogénation S.R voir alcanes
>C=C< H-X θ ordinaire >CH-CX< A.E
Markovnikov voir alcènes
>C=C< H-Br peroxydes
solv.apolaire >CH-CBr< effet
Kharasch A.R
anti-Markovnikov voir alcènes
R-OH H-X
PCl5
SOCl2
PBr3
pour HCl :
+ZnCl2
(Lucas)
R-X
RCl
RCl
RBr
Halogénation
• S.N
• réactivité :
HI>HBr>HCl
SOCl2 + pyridine →
inversion de configuration
SOCl2 + éther → rétention de
configuration
R-OH + H R-OH
H
R-X + H2O
X
R-OH + PCl5 → R-Cl + POCl3 + HCl
R-OH + SOCl2 → R-Cl + SO2 + HCl
R-OH + PBr3 → R-Br + P(OH)3
2- AROMATIQUES
Ar-H Cl2 , Br2cat AlCl3
ou FeBr3
Ar-Cl
Ar-Br S.E.Ar voir aromatiques
Ar-N+≡N
diazoïque X- : I-, Cl-
ou Br-cat : Cu2Cl2
pour Cl-
Cu2Br2
pour Br-
Ar-X
+ N2(g) Sandmeyer
(pour Cl et Br) Ar-NH2 + H+ + HNO2 → Ar-N+≡N + 2 H2O
Ar-N+≡N + X- → Ar-X + N2(g)
pour cette dernière réaction : mécanisme ionique pour I- et radicalaire pour Cl- et Br-
Ar-N+≡N BF4-∆ avec
précautions Ar-F + BF3réaction de
Schiemann Ar-NH2 + HNO2+ HBF4 BF4 + 2 H2O
-
Ar-N N
+
Ar-N N
+BF4
-∆Ar-F + BF3 + N2(g)

Réactif Conditions Produits Nom Remarques Mécanisme
ALCENE
X2 :Cl2,Br2,I2θ ambiante
solv : CCl4,éther CX-CX addition
électrophile A.E -antiaddition stéréospécifique
-réactivité : Cl2>Br2>I2
C=C + X2CC
X+ X-
C-C X
X+ énantiomère
HX :HCl,HBr,HI θ ambiante CH-CX A.E -réactivité :HI>HBr>HCl ;
-régiosélectivité :Markovnikov. C=C + H+C-C +
C-CX
H
H
X-
HXO :
HClO ;HBrO,HIO θ ambiante halohydrine :
CX-C-OH
A.E -antiaddition stéréospécifique ;
-régiosélectivité :addition de
l’électrophile X+
X-O-H + H X + H2O
++
C=C + X+CC
X
CC
X+ H2OC-C
X
OH
+ H+
+ énantiomère
H2O Catalyseur :
H2SO4
θ : 80 à 200°C
alcool
CH-C-OH
A.E -non stéréospécifique
-régioséléctivité : Markovnikov. C=C + H+C-CH
H2OC-CH + H+
OH
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
1
/
54
100%